General Information About Medulloblastoma and Other Central Nervous System (CNS) Embryonal Tumors
World Health Organization (WHO) Classification for CNS Embryonal Tumors and Pineoblastoma
Embryonal tumors are a collection of biologically heterogeneous lesions that share the tendency to disseminate throughout the nervous system via cerebrospinal fluid (CSF) pathways. Although there is significant variability, histologically these tumors are grouped together because they are at least partially composed of hyperchromatic cells (blue cell tumors on standard staining) with little cytoplasm, which are densely packed and demonstrate a high degree of mitotic activity. Other histological and immunohistochemical features, such as the degree of apparent cellular transformation along identifiable cell lineages (e.g., ependymal or glial), can be used to separate these tumors to some degree. However, a convention, which has been accepted by the WHO, also separates these tumors based on presumed location of origin within the CNS. Molecular studies have substantiated the differences between tumors arising in different areas of the brain and give partial credence to this classification approach.[1]
In 2016, the WHO proposed an integrated phenotypic and genotypic classification system for CNS tumors in which diagnoses are layered with WHO grade, histological classification, and molecular classification.[2] The term primitive neuroectodermal tumor (PNET) has been removed from the WHO diagnostic lexicon, although some rare entities (e.g., medulloepithelioma) have remained. A molecularly distinct entity, embryonal tumor with multilayered rosettes (ETMR), C19MC-altered, was added, encompassing embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma, and medulloepithelioma. The WHO classification will be updated as other molecularly distinct entities are defined.
The pathological diagnosis of embryonal tumors is based primarily on histological and immunohistological microscopic features. However, molecular genetic studies are employed increasingly to subclassify embryonal tumors. These molecular genetic findings are also being used for risk stratification and treatment planning.[3,4,5,6]
The 2021 WHO classification of embryonal tumors is as follows:[7,8]
-
Medulloblastoma.
- Medulloblastomas, molecularly defined.
- Medulloblastoma, WNT-activated.
- Medulloblastoma, SHH-activated and TP53-wild type.
- Medulloblastoma, SHH-activated and TP53-altered.
- Medulloblastoma, non-WNT/non-SHH.
- Medulloblastomas, histologically defined.
- Desmoplastic nodular medulloblastoma.
- Medulloblastoma with extensive nodularity.
- Large cell medulloblastoma.
- Anaplastic medulloblastoma.
- Other CNS embryonal tumors.
- Atypical teratoid/rhabdoid tumor.
- Cribriform neuroepithelial tumor.
- Embryonal tumor with multilayered rosettes.
- CNS neuroblastoma, FOXR2-activated.
- CNS tumor with BCOR internal tandem duplication.
- CNS embryonal tumor NEC/NOS.
Pineoblastoma was previously conventionally grouped with embryonal tumors. However, it is now categorized by the WHO as a pineal parenchymal tumor. The 2021 WHO classification of these tumors is as follows:[7,8]
- Pineocytoma.
- Pineal parenchymal tumor of intermediate differentiation.
- Pineoblastoma.
- Papillary tumor of the pineal region.
- Desmoplastic myxoid tumor of the pineal region, SMARCB1-altered.
Given that therapies for pineoblastomas are quite similar to those for embryonal tumors, pineoblastomas are discussed in this summary. A somewhat closely aligned tumor, pineal parenchymal tumor of intermediate differentiation (PPTID), has been identified but is not considered an embryonal tumor and primarily arises in adults.[2]
Anatomy
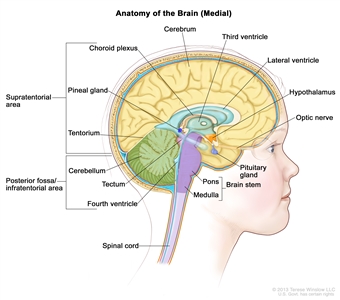
Figure 1. Anatomy of the inside of the brain, showing the pineal and pituitary glands, optic nerve, ventricles (with cerebrospinal fluid shown in blue), and other parts of the brain. The posterior fossa is the region below the tentorium, which separates the cortex from the cerebellum and essentially denotes the region containing the brain stem, cerebellum, and fourth ventricle.
Incidence
Embryonal tumors account for approximately 20% of primary CNS tumors (malignant CNS neoplasms and pilocytic astrocytomas) arising in children. These tumors occur along the pediatric age spectrum but tend to cluster early in life. The incidence of embryonal tumors in children aged 1 to 9 years is fivefold to tenfold higher than in adults (see Table 1).[9,10]
Table 1. Annual Incidence Rates for Childhood Central Nervous System Embryonal Tumors According to Agea
| Age Group (y) |
Annual Incidence Rate (Cases per 1 Million) |
|
a Source: National Childhood Cancer Registry.[9] |
| <5 |
10 |
| 5–9 |
7 |
| 10–19 |
2–3 |
Medulloblastomas comprise the vast majority of pediatric embryonal tumors. By definition, they arise in the posterior fossa (see Figure 1), where they constitute approximately 40% of all posterior fossa tumors. Other forms of embryonal tumors each make up 2% or less of all childhood brain tumors.
Diagnostic and Staging Evaluation
Imaging studies and CSF analysis are included in the diagnostic and staging evaluation.
Imaging studies
Diagnosis is usually made by either magnetic resonance imaging (MRI) or computed tomography (CT) scan. MRI is preferable because the anatomical relationship between the tumor and surrounding brain and tumor dissemination is better visualized with this method.[11,12]
After diagnosis, evaluation of embryonal tumors is quite similar, essentially independent of the histological subtype and the location of the tumor. Given the tendency of these tumors to disseminate throughout the CNS early in the course of illness, imaging evaluation of the neuraxis by MRI of the entire brain and spine is indicated. Preferably, this is done before surgery to avoid postoperative artifacts, especially blood. Such imaging can be difficult to interpret and must be performed in at least two planes, with and without the use of contrast enhancement (gadolinium).[13] A study of the significance of equivocal findings on spinal MRIs in children with medulloblastoma identified equivocal findings in 48 of 100 patients (48%). The study reported the following results:[14]
- Analysis by subgroup identified a higher proportion of equivocal findings in the SHH subgroup (P = .007).
- The 5-year overall survival (OS) rate in children with equivocal MRI findings (80%) was not different from the 5-year OS in patients who had normal MRI findings (84.8%), while OS in patients with M3 metastases was worse (54.7%) (P = .02).
In contrast, a Children's Oncology Group (COG) prospective study treated over 400 children without metastatic disease with a reduced dose (23.4 Gy) of craniospinal radiation therapy. Nearly 20% of patients with central neuroradiographic review were found to have either evidence of possible excessive residual disease and/or metastatic disease or were considered to have imaging inadequate to fully evaluate the neuroaxis. For patients with centrally reviewed imaging, children considered to have metastatic disease had poor OS compared with those with nondisseminated disease. The subgroup found to have inadequate imaging by central review had an intermediate survival rate between the children with adequate imaging and those who had metastatic disease.[13] In a subsequent prospective COG study that treated over 500 children with reduced-dose craniospinal radiation therapy (23.4 Gy or 18 Gy), patients with inadequate imaging had poorer survival.[15] Consensus guidelines for timing and neuroimaging techniques have been recommended and include details that outline standards for preoperative assessment of the entire neuroaxis and postoperative assessment of the amount of residual disease.[16]
After surgery, imaging of the primary tumor site is indicated to determine the extent of residual disease.
CSF analysis
After surgery, lumbar CSF analysis is performed, if deemed safe. Neuroimaging and CSF evaluation are considered complementary because as many as 10% of patients have evidence of free-floating tumor cells in the CSF without clear evidence of leptomeningeal disease on MRI scan.[17]
CSF analysis is conventionally done 14 to 21 days after surgery. If CSF is obtained within 14 days of the operation, detection of tumor cells within the spinal fluid is possibly related to the surgical procedure. In most staging systems, if fluid is obtained in the first few days after surgery and found to be positive for tumor cells, the positivity must be confirmed by a subsequent spinal tap to be considered diagnostically significant. In contrast, if CSF is negative for tumor cells at that time, then no confirmation is needed. When obtaining fluid by lumbar spinal tap is deemed unsafe, ventricular fluid can be obtained. However, this method may not be as sensitive as lumbar fluid assessment.[17]
Because embryonal tumors are very rarely metastatic to the bone, bone marrow, or other body sites at the time of diagnosis, studies such as bone marrow aspirates, chest x-rays, or bone scans are not indicated, unless there are symptoms or signs suggesting organ involvement.
Prognostic Factors
Various clinical and biological parameters have been associated with the likelihood of disease control of embryonal tumors after treatment.[4] Many of these factors have been shown to be predictive for medulloblastomas, although some are used to assign risk, to some degree, for other embryonal tumors. Parameters that are most frequently used to predict outcome include the following:[18,19]
It has become increasingly clear, especially for medulloblastomas, that outcome is also related to the molecular characteristics of the tumor, but this has not been definitively shown for other embryonal tumors.[1,5,6,20,21,22,23] OS rates range from 30% to 90%, depending on the molecular subtype of the medulloblastoma, extent of dissemination at time of diagnosis, and possibly other factors, such as the degree of resection. Children with medulloblastoma who survive for 5 years are considered cured of their tumor. Survival rates for other embryonal tumors are generally poorer, ranging from less than 5% to 50%. Specific survival rates are discussed within each subgroup in the summary.[24,25,26,27]
In older studies, the presence of brain stem involvement in children with medulloblastoma was found to be a prognostic factor. It has not been found to be of predictive value in subsequent studies that treated patients with both radiation and chemotherapy.[13,18]
An accurate diagnosis is critical for patients with embryonal tumors. For example, in the ACNS0332 (NCT00392327) trial that enrolled 80 patients with high-risk medulloblastoma, supratentorial CNS-PNET tumors, and pineoblastoma, 60 patients had sufficient tissue for evaluation. Thirty-one tumors were nonpineal in location, 22 (71%) of which represented tumors that were not intended for trial inclusion, including 18 high-grade gliomas, 2 atypical teratoid/rhabdoid tumors, and 2 ependymomas. Outcomes across tumor types were strikingly different. Patients with supratentorial embryonal tumors/pineoblastomas exhibited a 5-year event-free survival (EFS) rate of 62.8% (95% confidence interval [CI], 43.4%–82.2%) and an OS rate of 78.5% (95% CI, 62.2%–94.8%), whereas patients with molecularly classified high-grade gliomas had a 5-year EFS rate of 5.6% (95% CI, 0%–13%) and an OS rate of 12% (95% CI, 0%–24.7%). Survival rates for patients with high-grade gliomas were similar to those of patients who were enrolled in historical studies that avoided craniospinal irradiation and intensive chemotherapy. Thus, for patients with CNS-PNET/pineoblastoma, prognosis is considerably better than previously assumed when molecularly confirmed high-grade gliomas are removed.[28]
Prognosis is poor for patients with medulloepithelioma and ETMR, with 5-year survival rates ranging between 0% and 30%.[29,30,31] In a retrospective multivariate analysis of 38 patients, total or near-total resection, the use of radiation therapy, and the use of high-dose chemotherapy were associated with an improved prognosis.[32][Level of evidence C1] Another retrospective analysis included 159 patients with confirmed molecular diagnoses of primary ETMRs from the Rare Brain Tumor Registry (median age at diagnosis, 26 months; IQR, 18–36 months). The study revealed an EFS rate of 57% (95% CI, 47%–68%) at 6 months and 31% (95% CI, 21%–42%) at 2 years. The OS rate was 29% (95% CI, 20%–38%) at 2 years and 27% (95% CI, 18%–37%) at 4 years. OS was associated with nonmetastatic disease (hazard ratio [HR], 0.48; 95% CI, 0.28–0.80; P = .0057) and nonbrainstem location (HR, 0.42; 95% CI, 0.22–0.81; P = .013) on univariate analysis, as well as with gross-total resection (HR, 0.30; 95% CI, 0.16–0.58; P = .0014), use of high-dose chemotherapy (HR, 0.35; 95% CI, 0.19–0.67; P = .0020), and use of radiation therapy (HR, 0.21; 95% CI, 0.10–0.41; P < .0001) on multivariable analysis.[33][Level of evidence C1]
Extent of CNS disease at diagnosis
Patients with disseminated CNS disease at diagnosis are at highest risk of disease relapse.[17,18,19] Ten percent to 40% of patients with medulloblastoma have CNS dissemination at diagnosis. Infants have the highest incidence and adolescents and adults have the lowest incidence of CNS dissemination.
Nonmedulloblastoma embryonal tumors and pineoblastomas may also be disseminated at the time of diagnosis, although the incidence may be somewhat less than for medulloblastomas, with dissemination at diagnosis in approximately 10% to 20% of patients.[24,25] Patients with nonmedulloblastoma embryonal tumors and pineoblastomas who have disseminated disease at the time of diagnosis have a poor OS, with reported survival rates at 5 years ranging from 10% to 30%.[24,25,26,27,34]
Age at diagnosis
Age younger than 3 years at diagnosis portends an unfavorable outcome for those with medulloblastoma and, possibly, other embryonal tumors.[35,36,37,38,39,40] The exception is for those diagnosed with desmoplastic medulloblastoma/medulloblastoma with extensive nodularity (MBEN).
Amount of residual disease after definitive surgery
As a predictor of outcome, postoperative MRI measurement of the amount of residual disease after definitive surgery has been supplanted by extent of resection after surgery.[13]
In older studies, the extent of resection for medulloblastomas was found to be related to survival.[18,19,41,42] A Hirntumor and International Society of Paediatric Oncology study of 340 children reported that residual disease (>1.5 cm2) connoted a poorer 5-year EFS rate.[43] Extent of resection after surgery is still used to separate patients into risk groups, with patients having more than 1.5 cm2 of residual disease stratified into high-risk groups, with intensification of craniospinal irradiation to 36 Gy.
An international, retrospective, collaborative study included 787 patients of all ages with medulloblastoma who were treated in a variety of ways. The study incorporated molecular subgrouping and clinical factors in the analysis. The multivariate analysis found that subtotal resection (>1.5 cm2 residual), but not near-total resection (<1.5 cm2 residual), was associated with inferior progression-free survival compared with gross-total resection. This study suggested that attempts to completely remove the tumor, especially when the likelihood of neurological morbidity is high, are not warranted because there appears to be little or no benefit to gross-total resection when compared with near-total resection. It gives some credence to the present approach, in which patients with more than 1.5 cm2 of disease are considered higher-risk patients.[44] In a retrospective analysis of 1,100 patients with molecularly characterized medulloblastoma, subtotal resection was associated with worse survival in univariable analysis (P < .0001). However, subtotal resection was not independently prognostic in multivariable analyses and not prognostic in patients who did not have metastatic disease and received up-front craniospinal irradiation.[45] Prospective studies are needed to better define the impact of extent of resection on outcome within molecularly defined subgroups.
In patients with other forms of embryonal tumors, the extent of resection has not been definitively shown to impact survival.[26] However, in a COG study of 66 children with supratentorial embryonal tumors, extent of resection was found to be prognostic for those with localized disease at the time of diagnosis.[46]
Tumor histopathology
For medulloblastomas, histopathological features such as large cell variant, anaplasia, and desmoplasia have been shown in retrospective analyses to correlate with outcome.[36,47,48] In prospective studies, immunohistochemical and histopathological findings have not predicted outcome in children older than 3 years at diagnosis, with the exception of the large cell/anaplastic variant, which has been associated with poorer prognosis.[13,22,49] Several studies have observed that the histological finding of desmoplasia, seen in patients aged 3 years and younger with desmoplastic medulloblastoma, especially MBEN, connotes a significantly better prognosis compared with outcomes for infants and young children with classic or large cell/anaplastic medulloblastoma.[22,35,36,37,50]; [38][Level of evidence B4] Within the SHH group with TP53 variants, both somatic and constitutional (called Li-Fraumeni syndrome) TP53 variants may occur. Both of these variants connote a poor prognosis, compared with other SHH pathway–activated tumors.[23]
For other embryonal tumors, histological variations have not been associated with differing outcomes.
Biological/molecular tumor cell characteristics
Measure of minimal residual disease
In one study, CSF copy number variations, similar to those found in the primary tumors, were prognostic of relapse when present after radiation therapy or during or after chemotherapy. If this finding is replicated in prospective clinical trials and the technique becomes available, it will be an important measure of minimal residual disease and likely will become part of the baseline evaluation, as well as part of surveillance testing.[51]
Genomic analyses
For medulloblastoma, genomic analyses (including RNA gene expression and DNA methylation profiles, as well as DNA sequencing to identify variants) on both fresh-frozen and formalin-fixed, paraffin-embedded sections, have identified molecular subtypes.[3,4,5,6,20,21,52,53,54,55,56,57,58,59] These subtypes include those characterized by WNT pathway activation and SHH pathway activation, as well as additional subgroups characterized by MYC or MYCN alterations and other genomic alterations.[3,4,5,6,20,21,52,53,54,55,56,57,58] Children with medulloblastoma whose tumors show WNT pathway activation usually have an excellent prognosis. Within the non-WNT, non-SHH medulloblastoma group, there are subsets of patients with differing prognoses. For example, patients with chromosome 11 loss have an excellent prognosis, similar to those with WNT tumors.[15,60,61] Patients with SHH pathway–activated tumors have a prognosis that is influenced by the presence or absence of TP53 variants (favorable vs. unfavorable prognosis, respectively).[61] The outcome for the remaining patients is less favorable than for patients with WNT pathway activation. Variants in medulloblastoma are observed in a subtype-specific manner. CTNNB1 variants are observed in most WNT-subtype tumors. PTCH1, SMO, and SUFU variants are observed in the SHH-subtype tumors. The prognostic significance of recurring variants is closely aligned with that of the molecular subtype with which they are associated.[4,62] At recurrence, the subtype remains unchanged from the original molecular subtype at diagnosis.[63]
For nonmedulloblastoma embryonal tumors, integrative genomic analysis has also identified molecular subtypes with different outcomes. For more detailed information, see the Subtypes of nonmedulloblastoma embryonal tumors section.
Follow-Up After Treatment
Relapse in children with embryonal tumors is most likely to occur within the first 18 months of diagnosis.[43,64] Surveillance imaging of the brain and spine is usually undertaken at routine intervals during and after treatment (see Table 2). The frequency of such imaging, designed to detect recurrent disease at an early, asymptomatic state, has been arbitrarily determined and has not been shown to clearly influence survival.[65,66,67,68] Growth hormone replacement therapy has not been shown to increase the likelihood of disease relapse and should not impact the frequency of surveillance testing.[37]
Table 2. Surveillance Testing During and After Treatment for Medulloblastoma and Other Central Nervous System Embryonal Tumors
| Surveillance Period |
Frequency of Visits During Surveillance Period |
Testing |
| MRI = magnetic resonance imaging. |
|
a For pineoblastoma, continue spinal evaluations every 6 months until 5 years from diagnosis. Although these suggestions are based on a small sample size, there is evidence for continuing surveillance testing of the spine until 5 years after diagnosis.[69] |
| First 3 years after diagnosis |
Every 3 months |
Physical examination |
| Imaging of the brain every 3 months for the first 3 years, then every 6 months for the ensuing 2 years, and then as per preference of the treating physician or per protocol; MRI of the spine every 3 months for the first 2 years, then every 6 months for 1 year, and then as per preference of the treating physician or per protocola |
| Endocrinology evaluation once a year |
| Neuropsychological testing every 1–2 years |
| 3–5 years after diagnosis |
Every 6 months |
Physical examination |
| Imaging of the brain and spine once a year |
| Endocrinology evaluation once a year |
| Neuropsychological testing every 1–2 years |
| More than 5 years after diagnosis |
Once a year |
Physical examination |
| Imaging of the brain once a year |
| Endocrinology evaluation once a year |
| Neuropsychological testing every 1–2 years (optional) |
The development of surveillance strategies other than imaging for patients with medulloblastoma is the subject of ongoing research. In one study of 134 children with newly diagnosed medulloblastoma, copy number variants were detected at baseline in 123 patients (92%) by primary tumor testing and in 65 patients (49%) by CSF testing. Copy number variants were detected more frequently in the CSF of patients with disseminated disease and in those with subsequent progression. Prospective studies will be necessary to evaluate the potential for CSF copy number analysis to become a component of surveillance testing, as a measure of medulloblastoma minimal residual disease and early relapse.[51]
Childhood and adolescent cancer survivors require close monitoring because cancer therapy side effects may persist or develop months or years after treatment. For specific information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors, see Late Effects of Treatment for Childhood Cancer.
References:
-
Pomeroy SL, Tamayo P, Gaasenbeek M, et al.: Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature 415 (6870): 436-42, 2002.
-
Louis DN, Perry A, Reifenberger G, et al.: The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131 (6): 803-20, 2016.
-
Pfister S, Remke M, Benner A, et al.: Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci. J Clin Oncol 27 (10): 1627-36, 2009.
-
Taylor MD, Northcott PA, Korshunov A, et al.: Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123 (4): 465-72, 2012.
-
Kool M, Koster J, Bunt J, et al.: Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One 3 (8): e3088, 2008.
-
Ellison DW, Onilude OE, Lindsey JC, et al.: beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children's Cancer Study Group Brain Tumour Committee. J Clin Oncol 23 (31): 7951-7, 2005.
-
WHO Classification of Tumours Editorial Board, ed.: WHO Classification of Tumours: Central Nervous System Tumours. Vol. 6. 5th ed. IARC Press; 2021.
-
Louis DN, Perry A, Wesseling P, et al.: The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 23 (8): 1231-1251, 2021.
-
National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed February 25, 2025.
-
Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 30, 2024.
-
Chintagumpala MM, Paulino A, Panigrahy A, et al.: Embryonal and pineal region tumors. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Lippincott Williams and Wilkins, 2015, pp 671-99.
-
Jaju A, Li Y, Dahmoush H, et al.: Imaging of pediatric brain tumors: A COG Diagnostic Imaging Committee/SPR Oncology Committee/ASPNR White Paper. Pediatr Blood Cancer 70 Suppl 4 (Suppl 4): e30147, 2023.
-
Packer RJ, Gajjar A, Vezina G, et al.: Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol 24 (25): 4202-8, 2006.
-
Bennett J, Ashmawy R, Ramaswamy V, et al.: The clinical significance of equivocal findings on spinal MRI in children with medulloblastoma. Pediatr Blood Cancer 64 (8): , 2017.
-
Michalski JM, Janss AJ, Vezina LG, et al.: Children's Oncology Group Phase III Trial of Reduced-Dose and Reduced-Volume Radiotherapy With Chemotherapy for Newly Diagnosed Average-Risk Medulloblastoma. J Clin Oncol 39 (24): 2685-2697, 2021.
-
Warren KE, Vezina G, Poussaint TY, et al.: Response assessment in medulloblastoma and leptomeningeal seeding tumors: recommendations from the Response Assessment in Pediatric Neuro-Oncology committee. Neuro Oncol 20 (1): 13-23, 2018.
-
Fouladi M, Gajjar A, Boyett JM, et al.: Comparison of CSF cytology and spinal magnetic resonance imaging in the detection of leptomeningeal disease in pediatric medulloblastoma or primitive neuroectodermal tumor. J Clin Oncol 17 (10): 3234-7, 1999.
-
Zeltzer PM, Boyett JM, Finlay JL, et al.: Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the Children's Cancer Group 921 randomized phase III study. J Clin Oncol 17 (3): 832-45, 1999.
-
Yao MS, Mehta MP, Boyett JM, et al.: The effect of M-stage on patterns of failure in posterior fossa primitive neuroectodermal tumors treated on CCG-921: a phase III study in a high-risk patient population. Int J Radiat Oncol Biol Phys 38 (3): 469-76, 1997.
-
Thompson MC, Fuller C, Hogg TL, et al.: Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol 24 (12): 1924-31, 2006.
-
Northcott PA, Korshunov A, Witt H, et al.: Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29 (11): 1408-14, 2011.
-
Rutkowski S, von Hoff K, Emser A, et al.: Survival and prognostic factors of early childhood medulloblastoma: an international meta-analysis. J Clin Oncol 28 (33): 4961-8, 2010.
-
Kolodziejczak AS, Guerrini-Rousseau L, Planchon JM, et al.: Clinical outcome of pediatric medulloblastoma patients with Li-Fraumeni syndrome. Neuro Oncol 25 (12): 2273-2286, 2023.
-
Cohen BH, Zeltzer PM, Boyett JM, et al.: Prognostic factors and treatment results for supratentorial primitive neuroectodermal tumors in children using radiation and chemotherapy: a Childrens Cancer Group randomized trial. J Clin Oncol 13 (7): 1687-96, 1995.
-
Reddy AT, Janss AJ, Phillips PC, et al.: Outcome for children with supratentorial primitive neuroectodermal tumors treated with surgery, radiation, and chemotherapy. Cancer 88 (9): 2189-93, 2000.
-
Timmermann B, Kortmann RD, Kühl J, et al.: Role of radiotherapy in the treatment of supratentorial primitive neuroectodermal tumors in childhood: results of the prospective German brain tumor trials HIT 88/89 and 91. J Clin Oncol 20 (3): 842-9, 2002.
-
Jakacki RI, Zeltzer PM, Boyett JM, et al.: Survival and prognostic factors following radiation and/or chemotherapy for primitive neuroectodermal tumors of the pineal region in infants and children: a report of the Childrens Cancer Group. J Clin Oncol 13 (6): 1377-83, 1995.
-
Hwang EI, Kool M, Burger PC, et al.: Extensive Molecular and Clinical Heterogeneity in Patients With Histologically Diagnosed CNS-PNET Treated as a Single Entity: A Report From the Children's Oncology Group Randomized ACNS0332 Trial. J Clin Oncol : JCO2017764720, 2018.
-
Louis DN, Ohgaki H, Wiestler OD, et al.: The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114 (2): 97-109, 2007.
-
Sharma MC, Mahapatra AK, Gaikwad S, et al.: Pigmented medulloepithelioma: report of a case and review of the literature. Childs Nerv Syst 14 (1-2): 74-8, 1998 Jan-Feb.
-
Müller K, Zwiener I, Welker H, et al.: Curative treatment for central nervous system medulloepithelioma despite residual disease after resection. Report of two cases treated according to the GPHO Protocol HIT 2000 and review of the literature. Strahlenther Onkol 187 (11): 757-62, 2011.
-
Horwitz M, Dufour C, Leblond P, et al.: Embryonal tumors with multilayered rosettes in children: the SFCE experience. Childs Nerv Syst 32 (2): 299-305, 2016.
-
Khan S, Solano-Paez P, Suwal T, et al.: Clinical phenotypes and prognostic features of embryonal tumours with multi-layered rosettes: a Rare Brain Tumor Registry study. Lancet Child Adolesc Health 5 (11): 800-813, 2021.
-
Hansford JR, Huang J, Endersby R, et al.: Pediatric pineoblastoma: A pooled outcome study of North American and Australian therapeutic data. Neurooncol Adv 4 (1): vdac056, 2022.
-
Leary SE, Zhou T, Holmes E, et al.: Histology predicts a favorable outcome in young children with desmoplastic medulloblastoma: a report from the children's oncology group. Cancer 117 (14): 3262-7, 2011.
-
Giangaspero F, Perilongo G, Fondelli MP, et al.: Medulloblastoma with extensive nodularity: a variant with favorable prognosis. J Neurosurg 91 (6): 971-7, 1999.
-
von Bueren AO, von Hoff K, Pietsch T, et al.: Treatment of young children with localized medulloblastoma by chemotherapy alone: results of the prospective, multicenter trial HIT 2000 confirming the prognostic impact of histology. Neuro Oncol 13 (6): 669-79, 2011.
-
Rutkowski S, Gerber NU, von Hoff K, et al.: Treatment of early childhood medulloblastoma by postoperative chemotherapy and deferred radiotherapy. Neuro Oncol 11 (2): 201-10, 2009.
-
Rutkowski S, Bode U, Deinlein F, et al.: Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med 352 (10): 978-86, 2005.
-
Mynarek M, Pizer B, Dufour C, et al.: Evaluation of age-dependent treatment strategies for children and young adults with pineoblastoma: analysis of pooled European Society for Paediatric Oncology (SIOP-E) and US Head Start data. Neuro Oncol 19 (4): 576-585, 2017.
-
Albright AL, Wisoff JH, Zeltzer PM, et al.: Effects of medulloblastoma resections on outcome in children: a report from the Children's Cancer Group. Neurosurgery 38 (2): 265-71, 1996.
-
Taylor RE, Bailey CC, Robinson K, et al.: Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma: The International Society of Paediatric Oncology/United Kingdom Children's Cancer Study Group PNET-3 Study. J Clin Oncol 21 (8): 1581-91, 2003.
-
Lannering B, Rutkowski S, Doz F, et al.: Hyperfractionated versus conventional radiotherapy followed by chemotherapy in standard-risk medulloblastoma: results from the randomized multicenter HIT-SIOP PNET 4 trial. J Clin Oncol 30 (26): 3187-93, 2012.
-
Thompson EM, Hielscher T, Bouffet E, et al.: Prognostic value of medulloblastoma extent of resection after accounting for molecular subgroup: a retrospective integrated clinical and molecular analysis. Lancet Oncol 17 (4): 484-95, 2016.
-
Keeling C, Davies S, Goddard J, et al.: The clinical significance of sub-total surgical resection in childhood medulloblastoma: a multi-cohort analysis of 1100 patients. EClinicalMedicine 69: 102469, 2024.
-
Jakacki RI, Burger PC, Kocak M, et al.: Outcome and prognostic factors for children with supratentorial primitive neuroectodermal tumors treated with carboplatin during radiotherapy: a report from the Children's Oncology Group. Pediatr Blood Cancer 62 (5): 776-83, 2015.
-
McManamy CS, Lamont JM, Taylor RE, et al.: Morphophenotypic variation predicts clinical behavior in childhood non-desmoplastic medulloblastomas. J Neuropathol Exp Neurol 62 (6): 627-32, 2003.
-
Massimino M, Antonelli M, Gandola L, et al.: Histological variants of medulloblastoma are the most powerful clinical prognostic indicators. Pediatr Blood Cancer 60 (2): 210-6, 2013.
-
Eberhart CG, Kratz J, Wang Y, et al.: Histopathological and molecular prognostic markers in medulloblastoma: c-myc, N-myc, TrkC, and anaplasia. J Neuropathol Exp Neurol 63 (5): 441-9, 2004.
-
Garrè ML, Cama A, Bagnasco F, et al.: Medulloblastoma variants: age-dependent occurrence and relation to Gorlin syndrome--a new clinical perspective. Clin Cancer Res 15 (7): 2463-71, 2009.
-
Liu APY, Smith KS, Kumar R, et al.: Serial assessment of measurable residual disease in medulloblastoma liquid biopsies. Cancer Cell 39 (11): 1519-1530.e4, 2021.
-
Tabori U, Baskin B, Shago M, et al.: Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J Clin Oncol 28 (8): 1345-50, 2010.
-
Onvani S, Etame AB, Smith CA, et al.: Genetics of medulloblastoma: clues for novel therapies. Expert Rev Neurother 10 (5): 811-23, 2010.
-
Dubuc AM, Northcott PA, Mack S, et al.: The genetics of pediatric brain tumors. Curr Neurol Neurosci Rep 10 (3): 215-23, 2010.
-
Giangaspero F, Wellek S, Masuoka J, et al.: Stratification of medulloblastoma on the basis of histopathological grading. Acta Neuropathol 112 (1): 5-12, 2006.
-
Jones DT, Jäger N, Kool M, et al.: Dissecting the genomic complexity underlying medulloblastoma. Nature 488 (7409): 100-5, 2012.
-
Peyrl A, Chocholous M, Kieran MW, et al.: Antiangiogenic metronomic therapy for children with recurrent embryonal brain tumors. Pediatr Blood Cancer 59 (3): 511-7, 2012.
-
Kool M, Korshunov A, Remke M, et al.: Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 123 (4): 473-84, 2012.
-
Schwalbe EC, Williamson D, Lindsey JC, et al.: DNA methylation profiling of medulloblastoma allows robust subclassification and improved outcome prediction using formalin-fixed biopsies. Acta Neuropathol 125 (3): 359-71, 2013.
-
Shih DJ, Northcott PA, Remke M, et al.: Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol 32 (9): 886-96, 2014.
-
Goschzik T, Schwalbe EC, Hicks D, et al.: Prognostic effect of whole chromosomal aberration signatures in standard-risk, non-WNT/non-SHH medulloblastoma: a retrospective, molecular analysis of the HIT-SIOP PNET 4 trial. Lancet Oncol 19 (12): 1602-1616, 2018.
-
Polkinghorn WR, Tarbell NJ: Medulloblastoma: tumorigenesis, current clinical paradigm, and efforts to improve risk stratification. Nat Clin Pract Oncol 4 (5): 295-304, 2007.
-
Ramaswamy V, Remke M, Bouffet E, et al.: Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol 14 (12): 1200-7, 2013.
-
Packer RJ, Zhou T, Holmes E, et al.: Survival and secondary tumors in children with medulloblastoma receiving radiotherapy and adjuvant chemotherapy: results of Children's Oncology Group trial A9961. Neuro Oncol 15 (1): 97-103, 2013.
-
Shaw DW, Geyer JR, Berger MS, et al.: Asymptomatic recurrence detection with surveillance scanning in children with medulloblastoma. J Clin Oncol 15 (5): 1811-3, 1997.
-
Torres CF, Rebsamen S, Silber JH, et al.: Surveillance scanning of children with medulloblastoma. N Engl J Med 330 (13): 892-5, 1994.
-
Kramer ED, Vezina LG, Packer RJ, et al.: Staging and surveillance of children with central nervous system neoplasms: recommendations of the Neurology and Tumor Imaging Committees of the Children's Cancer Group. Pediatr Neurosurg 20 (4): 254-62; discussion 262-3, 1994.
-
Sabel M, Fleischhack G, Tippelt S, et al.: Relapse patterns and outcome after relapse in standard risk medulloblastoma: a report from the HIT-SIOP-PNET4 study. J Neurooncol 129 (3): 515-524, 2016.
-
Perreault S, Lober RM, Carret AS, et al.: Surveillance imaging in children with malignant CNS tumors: low yield of spine MRI. J Neurooncol 116 (3): 617-23, 2014.
Childhood Medulloblastoma
Hereditary Cancer Predisposition Syndromes Associated With Medulloblastoma
Increasingly, subsets of children with brain tumors, including medulloblastoma, have been found to have germline pathogenic or likely pathogenic variants, predisposing them to the development of medulloblastoma and other cancers.[1,2] These variants have obvious implications for the affected child, siblings, parents, and, potentially, other family members in regard to cancer surveillance, prevention, diagnosis, and management. The variants may also affect specific tumor treatment.
Medulloblastoma can arise in the setting of hereditary cancer predisposition syndromes in approximately 5% of patients.[1,2] A large study of over 1,000 patients demonstrated germline pathogenic variants in approximately 5% of all patients diagnosed with medulloblastoma. Germline pathogenic variants were identified in APC, BRCA2, PALB2, PTCH1, SUFU, and TP53.[2]
Syndromes known to be associated with medulloblastoma include the following:
- Turcot syndrome (related to germline pathogenic variants in APC),[3] exclusive to the WNT-activated subtype.[2]
- Rubinstein-Taybi syndrome (related to germline pathogenic variants in CREBBP).[4,5,6]
- Gorlin syndrome (also known as basal cell nevus syndrome or nevoid basal cell carcinoma syndrome, associated with germline PTCH1 and SUFU pathogenic variants).[7,8,9,10,11] The risk of developing medulloblastoma in patients with Gorlin syndrome appears to be higher in those with germline SUFU variants than in those with PTCH1 pathogenic variants. In one study, 2 of 115 individuals (1.7%) with Gorlin syndrome and a PTCH1 variant developed a pathology-proven medulloblastoma, compared with 3 of 9 individuals (33%) from three families with SUFU-related Gorlin syndrome. Each of the SUFU-related patients developed medulloblastoma before age 3 years.[11]
- Li-Fraumeni syndrome is related to germline pathogenic variants in TP53.[12,13] Germline TP53 pathogenic variants are restricted to the sonic hedgehog (SHH)–activated subtype.[2,14]
- Fanconi anemia (related to BRCA2 variants).[15,16,17,18]
Heterozygous deleterious germline pathogenic variants in GPR161 were identified in approximately 3% of cases of SHH medulloblastoma.[19]GPR161 is an inhibitor of SHH signaling. The median age at diagnosis for patients with GPR161 variants was 1.5 years. Loss of heterozygosity (LOH) at the GPR161 locus was noted in all tumors, with tumors from five of six patients showing copy-neutral LOH of chromosome 1q (on which GPR161 resides). The risk of nonmedulloblastoma cancers in patients with deleterious GPR161 variants is not defined.
Novel germline loss-of-function pathogenic variants in the largest subunit of the evolutionarily conserved Elongator complex, ELP1, were identified in 14% of pediatric patients with SHH medulloblastoma. ELP1 was the most common medulloblastoma predisposition gene, and it increased the prevalence of genetic predisposition to 40% among pediatric patients with SHH medulloblastoma.[20]
Sometimes medulloblastoma may be the initial manifestation of germline pathogenic variants in these predisposition genes. Germline testing should be considered in the following circumstances:
- APC variant testing in patients with WNT-activated medulloblastoma in the absence of a somatic CTNNB1 variant.
- SUFU, PTCH1, TP53, PALB2, and BRCA2 variant testing in patients with SHH-activated medulloblastoma. Patients with desmoplastic tumors with extensive nodularity should be carefully evaluated for stigmata of Gorlin syndrome.[7] One report observed that medulloblastoma with extensive nodularity (MBEN) was associated with Gorlin syndrome in 5 of 12 cases.[7] Gorlin syndrome is an autosomal dominant disorder in which those affected are predisposed to develop basal cell carcinomas later in life, especially in skin in the radiation portal. The syndrome can be diagnosed early in life by detection of characteristic dermatological and skeletal features such as keratocysts of the jaw, bifid or fused ribs, macrocephaly, and calcifications of the falx.[7]
- PALB2 and BRCA2 variant testing in patients with a family history of BRCA-associated cancers or homologous recombination repair deficiency.
Given the high frequency of underlying germline pathogenic or likely pathogenic variants associated with SHH medulloblastoma, all patients with this disease should be referred for genetic counseling.
Clinical Presentation
By definition, medulloblastomas arise in the posterior fossa.[21,22] In approximately 80% of children, medulloblastomas arise in the region of the fourth ventricle. Most of the early symptomatology is related to blockage of cerebrospinal fluid (CSF) and resultant accumulation of CSF in the brain, termed hydrocephalus. Children with medulloblastoma are usually diagnosed within 2 to 3 months of the onset of symptoms. Medulloblastoma commonly presents with the following signs and symptoms:[23]
- Relatively abrupt onset of headaches, especially in the morning on waking.
- Nausea and/or vomiting.
- Lethargy.
- Ataxia, including truncal unsteadiness.
- Some degree of nystagmus.
- Papilledema.
Twenty percent of patients with medulloblastoma will not have hydrocephalus at the time of diagnosis and are more likely to present initially with cerebellar deficits. For example, more laterally positioned medulloblastomas of the cerebellum may not result in hydrocephalus and, because of their location, are more likely to result in lateralizing cerebellar dysfunction (appendicular ataxia) manifested by unilateral dysmetria, unsteadiness, and weakness of the sixth and seventh nerves on the same side as the tumor. Later, as the tumor grows toward the midline and blocks CSF, the more classical symptoms associated with hydrocephalus become evident.
Cranial nerve findings are less common, except for unilateral or bilateral sixth nerve palsies, which are usually related to hydrocephalus.[23] At times, medulloblastomas will present explosively, with the acute onset of lethargy and unconsciousness resulting from hemorrhage within the tumor.
In infants, the presentation of medulloblastoma is more variable and may include the following:
- Nonspecific lethargy.
- Psychomotor delays.
- Loss of developmental milestones.
- Feeding difficulties.
- Increase in head circumference.
On examination, there may be bulging of the anterior fontanel due to increased intracranial pressure and abnormal eye movements, including eyes that are deviated downward (the so-called sun setting sign) because of loss of upgaze secondary to compression of the tectum of the midbrain.
Cellular and Molecular Classification
In the 2021 World Health Organization (WHO) classification, medulloblastoma is classified based on both histological and molecular features. The tumor is classified as medulloblastoma, histologically defined if no molecular testing has been performed.[22,24]
- Medulloblastoma.
- Medulloblastoma, molecularly defined.
- Medulloblastoma, WNT-activated.
- Medulloblastoma, SHH-activated and TP53-wild type.
- Medulloblastoma, SHH-activated and TP53-altered.
- Medulloblastoma, non-WNT/non-SHH.
- Medulloblastoma, histologically defined.
- Desmoplastic nodular medulloblastoma.
- Medulloblastoma with extensive nodularity.
- Large cell medulloblastoma.
- Anaplastic medulloblastoma.
Significant attention has been focused on medulloblastomas that display anaplastic features, including increased nuclear size, marked cytological pleomorphism, numerous mitoses, and apoptotic bodies.[25,26] Using the criteria of anaplasia is subjective because most medulloblastomas have some degree of anaplasia. Foci of anaplasia may appear in tumors with histological features of both classic and large cell medulloblastomas, and there is significant overlap between the anaplastic and large cell variants, which are frequently termed large cell/anaplastic medulloblastoma.[25,26] One convention is to consider medulloblastomas as anaplastic when anaplasia is diffuse (variably defined as anaplasia occurring in 50% to 80% of the tumor).
The incidence of medulloblastoma with the desmoplastic/nodular histological variant, which most commonly arises in a cerebellar hemisphere, is higher in infants, is less common in children, and increases again in adolescents and adults. The desmoplastic/nodular histological variant is different from MBEN. The nodular variant has an expanded lobular architecture. The MBEN subtype occurs almost exclusively in infants and generally carries an excellent prognosis.[7,27,28] However, a recent report used transcriptome sequencing to identify a subset of patients with MBENs that had a high frequency of germline pathogenic alterations in PTCH1 or SUFU. These patients had less favorable outcomes.[29]
Molecular subtypes of medulloblastoma
Multiple medulloblastoma subtypes have been identified by integrative molecular analysis.[30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53] Since 2012, the general consensus is that medulloblastoma can be molecularly separated into at least four core subtypes, including WNT-activated, sonic hedgehog (SHH)–activated, group 3, and group 4. In the 2021 World Health Organization (WHO) classification, the SHH subgroup has been divided into two groups based on TP53 status. Group 3 and group 4, which require methylation analysis for reliable separation, have been combined into medulloblastoma, non-WNT/non-SHH. Because the group 3 and group 4 terminology has been used extensively in completed studies and is still in use in ongoing and planned studies, this nomenclature will be maintained throughout the clinical discussion in this summary.[22,24]
Different regions of the same tumor are likely to have other disparate genetic variants, adding to the complexity of devising effective molecularly targeted therapy.[48] However, the major subtypes noted above remain stable across primary and metastatic components.[49,52]
Further subclassification within these subgroups is possible, which will provide even more prognostic information.[50,51,52]
Medulloblastoma, WNT-activated
WNT tumors are medulloblastomas with aberrations in the WNT signaling pathway and represent approximately 10% of all medulloblastomas.[50] WNT medulloblastomas show a WNT signaling gene expression signature and beta-catenin nuclear staining by immunohistochemistry.[54] They are usually histologically classified as classic medulloblastoma tumors and rarely have a large cell/anaplastic appearance. WNT medulloblastomas generally occur in older patients (median age, 10 years) and are infrequently metastasized at diagnosis. Recent studies have demonstrated the value of methylation profiling in identifying WNT-activated medulloblastomas. These studies included cases that would not be detected using other current testing methods (e.g., beta-catenin immunohistochemistry, CTNNB1 variant analysis, and evaluation for monosomy 6).[55]
CTNNB1 variants are observed in 85% to 90% of WNT medulloblastoma cases, with APC variants detected in many of the cases that lack CTNNB1 variants. Patients with WNT medulloblastoma whose tumors have APC variants often have Turcot syndrome (i.e., germline APC pathogenic variants).[51] In addition to CTNNB1 variants, WNT medulloblastoma tumors show 6q loss (monosomy 6) in 80% to 90% of cases. While monosomy 6 is observed in most medulloblastoma patients younger than 18 years at diagnosis, it appears to be much less common (approximately 25% of cases) in patients older than 18 years.[50,54]
The WNT subset is primarily observed in older children, adolescents, and adults and does not show a male predominance. The subset is believed to have brain stem origin, from the embryonal rhombic lip region.[56] WNT medulloblastomas are associated with a very good outcome in children, especially in individuals whose tumors have beta-catenin nuclear staining and proven 6q loss and/or CTNNB1 variants.[45,57,58,59] Retrospective studies have suggested that additional TP53 variants and OTX2 copy number gains may be associated with a worse prognosis for patients with WNT medulloblastoma.[60] These latter associations need to be verified in prospective studies.[61]
Medulloblastoma, SHH-activated andTP53-altered and medulloblastoma, SHH-activated andTP53-wild type
SHH tumors are medulloblastomas with aberrations in the SHH pathway and represent approximately 25% of medulloblastoma cases.[50] SHH medulloblastomas are characterized by chromosome 9q deletions; desmoplastic/nodular histology; and variants in SHH pathway genes, including PTCH1, PTCH2, SMO, SUFU, and GLI2.[54]
Heterozygous deleterious germline pathogenic variants in the GPR161 gene were identified in approximately 3% of cases of SHH medulloblastoma.[19]GPR161 is an inhibitor of SHH signaling. Median age at diagnosis for GPR161-altered cases was 1.5 years. Loss of heterozygosity (LOH) at the GPR161 locus was noted in all tumors, with tumors from five of six patients showing copy-neutral LOH of chromosome 1q (on which GPR161 resides).
Variants in the third nucleotide (r.3A>G) of the U1 spliceosomal small nuclear RNAs (snRNAs) are highly specific for SHH medulloblastoma.[62,63] U1 snRNA r.3A>G variants are observed in virtually all cases of SHH medulloblastoma in adults, in approximately one-third of cases in children and adolescents, and are absent in cases in infants.[63] U1 snRNA variants disrupt RNA splicing, leading to inactivation of tumor-suppressor genes (e.g., PTCH1) and activation of oncogenes (e.g., GLI2). The significance of U1 snRNA r.3A>G variants in specific SHH medulloblastoma subtypes is described below.
SHH medulloblastomas show a bimodal age distribution and are observed primarily in children younger than 3 years and in older adolescence/adulthood. The tumors are believed to emanate from the external granular layer of the cerebellum. The heterogeneity in age at presentation maps to distinctive subsets identified by further molecular characterization, as follows:
- The subset of medulloblastoma most common in children aged 3 to 16 years, termed SHH-alpha (a provisional subgroup in the 2021 medulloblastoma classification), is TP53 altered and is enriched for MYCN and GLI2 amplifications.[50,52] Amplifications of PTCH1 and SUFU may occur in this subtype and are mutually exclusive with TP53 variants (often germline), while the SMO variant is rare.[14,52,64] U1 snRNA variants occur in approximately 25% of SHH-alpha medulloblastoma cases and are associated with a very poor prognosis.[63]
- Two SHH subtypes that occur primarily in children younger than 3 years have been described.[50] One of these subtypes, termed SHH-1 (SHH-beta), is more commonly metastatic, with more frequent focal amplifications.[65] The second of these subtypes, termed SHH-2 (SHH-gamma), is enriched for the medulloblastoma with extensive nodularity (MBEN) histology. SHH pathway variants in children younger than 3 years with medulloblastoma include PTCH1 and SUFU variants.[52]SUFU variants are rarely observed in older children and adults, and they are commonly germline events.[64]
Reports that used DNA methylation arrays have also identified two subtypes of SHH medulloblastoma in young children.[28,65] One of the subtypes contained all of the cases with SMO variants, and it was associated with a favorable prognosis. The other subtype had most of the SUFU variants, and it was associated with a much lower progression-free survival (PFS) rate. PTCH1 variants were present in both subtypes.
- A fourth SHH subtype, termed SHH-delta, includes most of the adult cases of SHH medulloblastoma.[50] Virtually all cases of SHH-delta medulloblastoma have the U1 snRNA r.A>3 variant,[63] and approximately 90% of cases have TERT promoter variants.[50]PTCH1 and SMO variants are also observed in adults with SHH medulloblastoma.
The outcome for patients with nonmetastatic SHH medulloblastoma is relatively favorable for children younger than 3 years and for adults.[50] Young children with the MBEN histology have a particularly favorable prognosis.[7,27,66,67,68] Patients with SHH medulloblastoma at greatest risk of treatment failure are children older than 3 years whose tumors have TP53 variants, often with co-occurring GLI2 or MYCN amplification and large cell/anaplastic histology.[50,64,69]
Patients with unfavorable molecular findings have an unfavorable prognosis, with fewer than 50% of patients surviving after conventional treatment.[46,64,69,70,71,72]
The 2021 WHO classification identifies SHH medulloblastoma with a TP53 variant as a distinctive entity (medulloblastoma, SHH-activated and TP53-altered).[22,24] Approximately 25% of SHH-activated medulloblastoma cases have TP53 variants, with a high percentage of these cases also showing a TP53 germline pathogenic variant (9 of 20 in one study). These patients are commonly between the ages of 5 years and 18 years and have a worse outcome (5-year overall survival rate, <30%).[71] The tumors often show large cell anaplastic histology.[71] A larger retrospective study has confirmed the poor prognosis of these patients.[14]
Medulloblastoma, non–WNT/non–SHH-activated
The WHO classification combines group 3 and group 4 medulloblastoma cases into a single entity, partly based on the absence of immediate clinical impact for this distinction. Group 3 represents approximately 25% of medulloblastoma cases, while group 4 represents approximately 40% of medulloblastoma cases.[50,54] Both group 3 and group 4 medulloblastoma patients are predominantly male.[39,52] Group 3 and group 4 medulloblastomas can be further subdivided based on characteristics such as gene expression and DNA methylation profiles, but the optimal approach to their subdivision is not established.[50,51]
Various genomic alterations are observed in group 3 and group 4 medulloblastomas. However, no single alteration occurs in more than 10% to 20% of cases. Genomic alterations include the following:
- MYC amplification was the most common distinctive alteration reported for group 3 medulloblastoma, occurring in approximately 15% of cases.[44,51]
- The most common distinctive genomic alteration described for group 4 medulloblastoma (observed in approximately 15% of cases) was activation of PRDM6 by enhancer hijacking, resulting from the tandem duplication of the adjacent SNCAIP gene.[51]
- Other genomic alterations were observed in both group 3 and group 4 cases, including MYCN amplification and structural variants leading to GFI1 or GFI1B overexpression through enhancer hijacking.
- Isochromosome 17q (i17q) is the most common cytogenetic abnormality and is observed in a high percentage of group 4 cases, as well as in group 3 cases, but it is rarely observed in WNT and SHH medulloblastoma.[44,51] Prognosis for group 3 and group 4 patients does not appear to be affected by the presence of i17q.[73]
Group 3 patients with MYC amplification or MYC overexpression have a poor prognosis.[52] Fewer than 50% of these patients survive 5 years after diagnosis.[50] This poor prognosis is especially true in children younger than 4 years at diagnosis.[46] However, patients with group 3 medulloblastoma without MYC amplification who are older than 3 years have a prognosis similar to that of most patients with non-WNT medulloblastoma, with a 5-year PFS rate higher than 70%.[70,73]
Group 4 medulloblastomas occur throughout infancy and childhood and into adulthood. The prognosis for group 4 medulloblastoma patients is similar to that for patients with other non-WNT medulloblastomas. Prognosis may be affected by additional factors such as the presence of metastatic disease, chromosome 11q loss, and chromosome 17p loss.[43,44,50,69] One study found that group 4 patients with either chromosome 11 loss or gain of chromosome 17 were low risk, regardless of metastases. In cases lacking both of these cytogenetic features, metastasis at presentation differentiated between high and intermediate risk.[69]
For group 3 and group 4 standard-risk patients (i.e., without MYC amplification or metastatic disease), the gain or loss of whole chromosomes appears to connote a favorable prognosis. This finding was derived from the data of 91 patients with non-WNT/non-SHH medulloblastoma enrolled in the SIOP-PNET-4 (NCT01351870) clinical trial and was confirmed in an independent group of 70 children with non-WNT/non-SHH medulloblastoma treated between 1990 and 2014.[73] Chromosomal abnormalities include the following:
- The gain/loss of one or more whole chromosomes was associated with a 5-year event-free survival (EFS) rate of 93%, compared with 64% for no whole chromosome gains/losses.
- The most common whole chromosomal gains/losses are gain of chromosome 7 and loss of chromosomes 8 and 11.
- The optimally performing prognosis discriminator was determined to be the occurrence of two or more of the following aberrations: chromosome 7 gain, chromosome 8 loss, and chromosome 11 loss. Approximately 40% of group 3 and group 4 standard-risk patients had two or more of these chromosomal aberrations and had a 5-year EFS rate of 100%, compared with 68% for patients with fewer than two aberrations.
- In an independent cohort, the prognostic significance of two or more gains/losses versus zero or one gain/loss of chromosomes 7, 8, and 11 was confirmed (5-year EFS rate, 95% for patients with two or more vs. 59% for patients with one or fewer).
The classification of medulloblastoma into the four major subtypes will likely be altered in the future.[50,51,72,74,75] Further subdivision within subgroups based on molecular characteristics is likely because each of the subgroups is further molecularly dissected, although the studies are nearing consensus as data from multiple independent studies are merged. As an example, using complementary bioinformatics approaches, concordance was analyzed among multiple large, published cohorts, and a more unified subgrouping was described. For children with group 3 and group 4 medulloblastomas, eight distinct subgroups were determined by DNA methylation clustering. Specific subgroups had different prognoses.[43,54,64,76]
It is unknown whether the classification for adults with medulloblastoma has a predictive ability similar to that for children.[44,46] In one study of adult patients with medulloblastoma, MYC oncogene amplifications were rarely observed, and tumors with 6q deletion and WNT activation (as identified by nuclear beta-catenin staining) did not share the excellent prognosis seen in pediatric medulloblastomas. However, another study did confirm an excellent prognosis for WNT-activated tumors in adults.[44,46]
Staging Evaluation
Historically, staging was based on an intraoperative evaluation of both the size and extent of the tumor, coupled with postoperative neuroimaging of the brain and spine and cytological evaluation of CSF (the Chang system). Intraoperative evaluation of the extent of the tumor has been supplanted by neuraxis imaging before diagnosis and postoperative imaging to determine the amount of primary site residual disease. The following tests and procedures are now used for staging:
- Magnetic resonance imaging (MRI) of the brain and spine (often done preoperatively).
- Postoperative MRI of the brain to determine the amount of residual disease.
- Lumbar CSF analysis.[77,78,79]
The tumor extent is defined as:
- M0: No dissemination.
- M1: CSF-positive cytology only.
- M2: Gross nodular seeding in cerebellar-cerebral subarachnoid space and/or lateral or third ventricle.
- M3: Gross nodular seeding in spinal subarachnoid space.
- M4: Extraneural metastasis.
Postoperative degree of residual disease is designated as:
- Gross-total resection/near-total resection: No or minimal (≤1.5 cm2) evidence of residual disease after resection.
- Subtotal resection: Residual disease after diagnosis (>1.5 cm2 of measurable residual disease).
- Biopsy: No tumor resection; only a sample of tumor tissue is removed.
Since the 1990s, prospective studies have been performed using this staging system to separate patients into average-risk and high-risk medulloblastoma subgroups.[78,79,80]
The presence of diffuse (>50% of the pathological specimen) histological anaplasia has been added to staging systems. If diffuse anaplasia is found, patients with otherwise average-risk disease are upstaged to high-risk disease.
Risk Stratification
Risk stratification is based on neuroradiographic evaluation for disseminated disease, CSF cytological examination, postoperative neuroimaging evaluation for the amount of residual disease, and patient age. For more information, see the Staging Evaluation section. Patients older than 3 years with medulloblastoma have been stratified into the following two risk groups:
-
Average risk: Children older than 3 years with tumors that are totally resected or near-totally resected (≤1.5 cm2 of residual disease) and who have no metastatic disease.[78]
-
High risk: Children older than 3 years with metastatic disease and/or subtotal resection (>1.5 cm2 of residual disease).[78] Metastatic disease includes neuroradiographic evidence of disseminated disease, positive cytology in lumbar or ventricular CSF obtained more than 14 days after surgery, or extraneural disease.[78] Children with tumors showing diffuse anaplasia and who otherwise would be considered average risk are assigned to the high-risk group.[26,38]
For younger children (younger than 3 years in some studies and younger than 4 or 5 years in others), similar separation into average-risk (no dissemination and ≤1.5 cm2 of residual disease) or high-risk (disseminated disease and/or >1.5 cm2 of residual disease) groups has been used. Histological findings of desmoplasia have also been used to connote a more favorable risk subgrouping, especially for the MBEN subgroup.[81,82]
Assigning a risk group based on the extent of resection and disease at diagnosis may not predict treatment outcome. Molecular genetics and histological factors may be more informative, although they must be evaluated in the context of patient age, the extent of disease at the time of diagnosis, and treatment received.[43,72,83] The risk characterizations of molecular subdivisions are changing and are becoming integrated into risk stratification schema to assign treatment in North American prospective studies (e.g., NCT01878617 and NCT02724579).[74]
Treatment Option Overview for Childhood Medulloblastoma
Table 3 describes the standard treatment options for newly diagnosed and recurrent childhood medulloblastoma.
Surgery
Surgery is considered a standard part of treatment for histological confirmation of tumor type and as a means to improve outcome. Total or near-total resections are considered optimal if they can be performed safely.[84,85]
Postoperatively, children may have significant neurological deficits caused by preoperative tumor-related brain injury, hydrocephalus, or surgery-related brain injury.[86][Level of evidence C1] A significant number of patients with medulloblastoma develop cerebellar mutism syndrome (also known as posterior fossa syndrome). Symptoms of cerebellar mutism syndrome, which usually appear within 1 or 2 days after surgery, include the following:
- Loss of speech.
- Suprabulbar palsies.
- Ataxia.
- Hypotonia.
- Emotional lability.
The etiology of cerebellar mutism syndrome remains unclear, although cerebellar vermian damage and/or disruption of cerebellar-cortical tracts has been postulated as the possible cause of the mutism.[87,88]; [89][Level of evidence C1] In two Children's Cancer Group studies that evaluated children with both average-risk and high-risk medulloblastoma, the syndrome was identified in nearly 25% of patients.[88,89,90]; [91][Level of evidence C1] A retrospective analysis of 370 patients with medulloblastoma identified younger age, larger tumor size, and midline tumor location as risk factors for developing mutism. This finding is consistent with previous observations. Investigators also observed a correlation between medulloblastoma subtype and risk of mutism. Mutism was more common in patients with group 3 and group 4 medulloblastomas (30%–35% of patients) and less frequent in children with SHH-activated tumors (7% of patients).[92] A prospective study that included longitudinal neurological examination of 178 patients with medulloblastoma identified similar risk factors for mutism (higher risk with younger age; lower risk with SHH subtype), most likely because SHH-activated tumors tend to be located in the hemispheres and not in the midline. The study also reported a higher risk of developing mutism in patients who undergo tumor resections at low-volume surgery centers.[93] Approximately 50% of patients with this syndrome manifest long-term, permanent neurological and neurocognitive sequelae.[89,91]
Radiation therapy
Radiation therapy to the primary tumor site is usually in the range of 54 Gy to 55.8 Gy.[94] In most instances, this therapy is given with a margin of 1 cm to 2 cm around the primary tumor site, preferably by conformal techniques.[94] Reducing boost volumes for the whole posterior fossa and to the tumor bed plus margins did not compromise outcomes in average-risk patients in the Children's Oncology Group (COG) ACNS0331 (NCT00085735) study.[59][Level of evidence A1] For all medulloblastomas in children older than 3 or 4 years at diagnosis, craniospinal radiation therapy is given at doses ranging between 23.4 Gy and 36 Gy, depending on risk factors such as extent of disease at diagnosis. A prospective phase II toxicity study of proton radiation therapy [95] and a retrospective efficacy report of protons versus photons for medulloblastoma [96] demonstrated equivalent outcomes for PFS, overall survival (OS), patterns of relapse, and delayed toxic effects. A retrospective study of 84 children who received either proton (n = 38) or photon (n = 46) radiation therapy demonstrated similar rates of grade 3 and grade 4 ototoxicity despite low mean cochlear doses in children who received proton radiation therapy, suggesting that other factors (e.g. cisplatin, initial tumor location in relationship to the vestibulocochlear nerve [eighth cranial nerve]) contribute to ototoxicity.[97] The comparative outcomes of these treatment technologies are under investigation.
Chemotherapy is usually administered during and after radiation therapy.
For children younger than 3 years, efforts are made to omit or delay radiation therapy, given the profound impact of radiation at this age. Children of all ages are susceptible to the adverse effects of radiation on brain development. Debilitating effects on neurocognitive development, growth, and endocrine function have been frequently observed, especially in younger children.[98,99,100,101,102]
Chemotherapy
Chemotherapy, usually given during and after radiation therapy, is a standard component of treatment for older children with medulloblastoma and other embryonal tumors. Chemotherapy can be used to delay and sometimes obviate the need for radiation therapy in 20% to 40% of children younger than 3 to 4 years with nondisseminated medulloblastoma.[103,104]; [102][Level of evidence C1]
Treatment of Childhood Medulloblastoma
Treatment of younger children with medulloblastoma
The 5-year event-free survival (EFS) rates for young children with medulloblastoma, arbitrarily described in the past as aged 3 years and younger at diagnosis, have ranged between 30% and 70%. There is no consensus as to what age constitutes a younger population of children with medulloblastoma who are best treated with immediate postsurgery chemotherapy and delayed or no radiation therapy. Most studies agree that in patients aged 3 years and younger, initial chemotherapy should be strongly considered. In patients between the ages of 3 and 4 years, and possibly as old as age 5 years, some investigators have recommended that radiation therapy be delayed or omitted entirely. Such decisions are based on multiple factors, including histological subtype, molecular subtype, extent of disease at diagnosis, preexisting neurological and neurodevelopmental status, and family preferences. Most long-term survivors who have been successfully treated with chemotherapy alone have had nondisseminated completely resected tumors.[81,103,105]; [106][Level of evidence B4] Several studies that have used chemotherapy alone for younger patients have observed that the finding of desmoplasia (seen in patients with desmoplastic medulloblastoma or MBEN) and/or molecular evidence of SHH signaling suggests a significantly better prognosis than the finding of classic or large cell/anaplastic medulloblastoma.[7,27,66,67,68]; [82][Level of evidence B4]
The treatment of younger children with newly diagnosed medulloblastoma continues to evolve. Results have been variable, and comparison across studies has been difficult because of differences in the drug regimens used and the utilization of craniospinal and local boost radiation therapy at the end of chemotherapy or when children reached age 3 years in some studies.
Standard treatment options for younger children with newly diagnosed medulloblastoma include the following:
-
Surgery.
-
Adjuvant chemotherapy.
Surgery
If feasible, complete surgical resection of the tumor is the optimal treatment. Surgical resectability is associated with histology, as patients with desmoplastic/nodular medulloblastoma or MBEN have a higher rate of complete resection than patients with classic medulloblastoma.[67,68]
Adjuvant chemotherapy
Therapies for younger children with medulloblastoma have included the use of multiagent chemotherapeutic approaches, including drugs such as cyclophosphamide, etoposide, cisplatin, and vincristine, with or without concomitant high-dose intravenous and/or intraventricular methotrexate.[68,81,103,105,107,108]; [109,110][Level of evidence B4] The efficacy of chemotherapy has varied, depending on the histology and/or molecular subtype of the tumor.
Desmoplastic/MBEN medulloblastoma and/or tumors with SHH signaling
A series of studies have demonstrated that intensive chemotherapy, including either high-dose systemic and intraventricular methotrexate or high-dose chemotherapy supported by stem cell rescue, without radiation therapy, is an effective treatment for most infants and very young children with medulloblastoma.
Evidence (chemotherapy):
- In the German Hirntumor (HIT) 2000 multicenter trial, a multiagent chemotherapy regimen that included high-dose intravenous and intraventricular methotrexate was used. This drug regimen did not include high-dose chemotherapy supported by bone marrow or peripheral stem cell rescue.[81]
- Nineteen patients with desmoplastic medulloblastoma or MBEN had a 5-year EFS rate of 90% (±7%) and an OS rate of 100% (±0%).
- All patients were treated with postoperative chemotherapy alone, and no radiation was given before progression.
- An expanded cohort of the German HIT 2000 trial included an additional 23 children with nodular desmoplasia or MBEN who were treated with the same regimen. The following results were reported:[111]
- Combined results confirmed the excellent survival, with a 5-year EFS rate of greater than 90%.
- In this expanded cohort, molecular characterization was performed and a subset of tumors with SHH signaling were identified. These patients with tumors demonstrating SHH signaling had a similar excellent prognosis.
- Further characterization of the SHH signaling molecular subtype by chromosomal aberrations did not identify any differences in EFS or OS.
- Other studies have suggested that further subdivision by chromosomal aberrations in young children with SHH-driven medulloblastoma was predictive of outcome.
- A COG clinical trial (CCG-9921) also had a favorable outcome for children with desmoplastic medulloblastoma (including MBEN). In this study, patients with desmoplastic tumors did not receive radiation therapy before progression.[103]
- Patients in the desmoplastic group achieved an EFS rate of 77% (±9%) and an OS rate of 85% (±8%), compared with an EFS rate of 17% (±5%) and an OS rate of 29% (±6%) for patients in the nondesmoplastic group (P < .0001 for both EFS and OS comparisons).
- The COG-ACNS1221 (NCT02017964) study tested systemic chemotherapy that was identical to the chemotherapy used in the German HIT 2000 trial, except for the omission of intraventricular methotrexate.[28]
- The study was closed early because of a higher-than-expected rate of relapse, with a 2-year PFS rate of 52% in the 25 patients who were studied.
- Another treatment option for children younger than 3 years at diagnosis is high-dose chemotherapy. Results of trials using higher-dose, marrow-ablative chemotherapeutic regimens supported by stem cell rescue have also demonstrated that a subgroup of patients with medulloblastoma who are younger than 3 years and, in some studies, younger than 5 years at the time of diagnosis can be successfully treated with chemotherapy alone.[104,106,112][Level of evidence B4]; [113]
- The best survival rates using this higher-dose chemotherapy approach have been seen in patients with desmoplastic medulloblastoma and MBEN.[113]
- After treatment with chemotherapy without concomitant radiation therapy, patients with nondisseminated disease have achieved survival rates of nearly 90%, and patients with disseminated disease have achieved survival rates of 80%.
- One study reported the outcomes of infants and young children with relapsed medulloblastoma who were initially treated without craniospinal irradiation (CSI).[114]
- A substantial portion of these children were treated with CSI-based regimens and their disease was successfully salvaged.
- The 3-year postrelapse survival rate was 52.4% for patients treated with curative intent.
- The report found that older age at diagnosis, local relapse, and the SHH infant medulloblastoma subtype were associated with better postrelapse survival.
- The addition of chemotherapy to CSI did not improve outcomes.
Nondesmoplastic, non-MBEN, and non-SHH signaling–driven medulloblastoma
Compared with children with desmoplastic medulloblastoma or MBEN treated with current intensive chemotherapy regimens, children with other histological and/or molecular subtypes fare less well. One study suggested that patients with molecularly identified group 4 tumors did well with chemotherapy alone.[111]
Evidence (chemotherapy):
- In children with nondesmoplastic, non-MBEN, and/or non-SHH–signaling tumors, the EFS rates are less than 40% despite the use of intensive chemotherapy supplemented with methotrexate (intravenous and intraventricular) or the use of high-dose chemotherapy regimens supported with stem cell rescue.[68,103,111,113,115]
- Outcome is particularly poor when these patients have disseminated disease.
- In some studies, radiation therapy to the primary tumor site and/or craniospinal axis has been added after chemotherapy, which makes the assessment of the efficacy of chemotherapy more difficult.[111,113,115]
There is no consensus on how much radiation therapy (dose and extent) should be given and at what age radiation therapy should be instituted in young patients with localized or disseminated disease.
- In the expanded HIT 2000 study, the addition of focal radiation therapy to the primary tumor site in patients with localized disease after chemotherapy did not improve EFS or OS.[111]
- In the St. Jude Children's Research Hospital (SJCRH) SJYC07 (NCT00602667) study, focal radiation therapy also did not improve EFS in infants with medulloblastoma denoted as intermediate risk (5-year EFS rate, 25% ± 12%).[65]
- The COG P9934 (NCT00006461) study, which also employed focal radiation therapy, had a similar EFS (4-year EFS rate, 23% ± 12%) for patients with nondesmoplastic medulloblastoma.[116]
- In the SJCRH SJYC07 study, 29 of the 54 infants with medulloblastoma whose disease progressed received CSI (median dose, 36 Gy). Of the 29 patients, 18 (62%) survived, compared with 6 of 25 patients (24%) who did not receive CSI.[65]
Treatment of children older than 3 years with average-risk medulloblastoma
Standard treatment options for children older than 3 years with newly diagnosed average-risk medulloblastoma include the following:
-
Surgery.
-
Adjuvant radiation therapy.
-
Adjuvant chemotherapy.
Surgery
If feasible, total or near-total removal of the tumor is considered optimal.[84]
Adjuvant radiation therapy
Radiation therapy is usually initiated after surgery with or without concurrent chemotherapy.[117,118,119] The best survival results for children with medulloblastoma have been obtained when radiation therapy is initiated within 4 to 6 weeks postsurgery.[118,119,120]; [94,121][Level of evidence A1] A pilot study in children with WNT-activated medulloblastoma attempted to omit craniospinal radiation therapy (and treat patients with postsurgical chemotherapy alone). The study was aborted after the first two patients had early tumor recurrences.[122]
The radiation dose for patients with average-risk medulloblastoma is 54 Gy to the posterior fossa or local tumor bed and 23.4 Gy to the entire neuraxis (i.e., the whole brain and spine), termed CSI.[117,118,119,123]
Evidence (adjuvant radiation therapy):
- With radiation therapy alone, using a craniospinal radiation dose of 35 Gy with a boost to the posterior fossa of 55 Gy, 5-year EFS rates range between 50% and 65% in patients with nondisseminated disease.[80,118]
- The minimal dose of craniospinal radiation needed for disease control is unknown. Attempts to lower the dose of craniospinal radiation therapy to 23.4 Gy without chemotherapy have resulted in an increased incidence of isolated leptomeningeal relapse.[123] One series attempted to treat children with WNT-activated tumors using focal radiation therapy alone, without CSI. The study showed an unacceptable incidence of neuroaxial failure with the omission of up-front CSI.[124]
Lower doses and boost volume of craniospinal radiation were evaluated in a COG study (NCT00085735). Children aged 3 to 7 years were randomly assigned to receive a craniospinal radiation dose of either 18 Gy or 23.4 Gy, as well as whole posterior fossa versus limited target volume boost to the tumor bed.[59][Level of evidence A1]
- Results revealed that 18 Gy of CSI was inferior to 23.4 Gy of CSI (5-year EFS rate of 82.6% ± 4.2% and OS rate of 85.8% for patients who received 23.4 Gy vs. EFS rate of 71.9% ± 4.9% and OS rate of 77.9% ± 4.9% for patients who received 18 Gy).
- The 5-year EFS rate was 82.5% for patients who received radiation therapy targeting the tumor bed, compared with 80.5% for patients who received posterior fossa radiation therapy. Therefore, radiation therapy targeting the tumor bed was not inferior to posterior fossa radiation therapy (hazard ratio, 0.97; 94% upper confidence interval, 1.32).
Analysis according to molecular subgroups demonstrated that children with group 4 medulloblastoma who received 18 Gy of craniospinal radiation therapy had poorer EFS than those who received 23.4 Gy. This was not demonstrated in the other molecular subgroups, although the study was not sized for molecular subgroup analysis.[59] Craniospinal radiation dose reduction to 18 Gy is currently being investigated in WNT medulloblastoma patients (NCT02724579), the molecular subgroup with the best prognosis.
- The SIOP-PNET-4 (NCT01351870) study compared daily radiation therapy (1.8 Gy fractions with 23.4 Gy to the neuraxis and a 30 Gy boost to the posterior fossa) with twice-per-day radiation (1 Gy fractions with 36 Gy and a 24-Gy boost to the posterior fossa).[125]
- With a median follow-up of 7.8 years, the 10-year OS was not significantly different between the two radiation groups.
- Long-term side effects were not reported in this study.
- If chemotherapy is added after radiation therapy, 23.4 Gy of craniospinal radiation therapy has been shown to be an effective dose.[94,125,126,127] Lower doses are being evaluated.
- Although the standard boost in medulloblastoma is to the entire posterior fossa, failure data patterns reveal that radiation therapy to the tumor bed instead of the entire posterior fossa is equally effective and may be associated with reduced toxicity.[128,129]; [59][Level of evidence A1]
Adjuvant chemotherapy
Chemotherapy is now a standard component of the treatment of children with average-risk medulloblastoma.
Evidence (adjuvant chemotherapy):
- Prospective randomized trials and single-arm trials suggest that adjuvant chemotherapy given during and after radiation therapy improves OS for children with average-risk medulloblastoma.[91,117,118,119,120,121]
- Radiation therapy and chemotherapy given during and after surgery has demonstrated 5-year EFS rates of 70% to 85%.[117,118,119]; [130][Level of evidence B4]
- A lower radiation dose of 23.4 Gy to the neuraxis when coupled with chemotherapy has been shown to result in disease control in up to 85% of patients and may decrease the severity of long-term neurocognitive sequelae.[94,126,127,131]
- A variety of chemotherapeutic regimens have been successfully used, including the combination of cisplatin, lomustine, and vincristine or the combination of cisplatin, cyclophosphamide, and vincristine.[117,118,131,132]
- These therapies have increased 5-year and 10-year EFS and OS rates and have likely decreased the incidence of late relapse.
- However, long-term survivors treated with multimodality therapy are at a high risk of late effects such as hearing loss, cardiac complications, and secondary neoplasms.[133]
In addition, postradiation high-dose cyclophosphamide supported by peripheral stem cell rescue, but with reduced cumulative doses of vincristine and cisplatin, has resulted in similar survival rates.[58]
- Although medulloblastoma is often sensitive to chemotherapy, preradiation chemotherapy has not been shown to improve survival in patients with average-risk medulloblastoma, compared with radiation therapy and subsequent chemotherapy. In some prospective studies, preradiation chemotherapy has been related to a poorer rate of survival.[118,119,120,121]
Treatment of children older than 3 years with high-risk medulloblastoma
Standard treatment options for children older than 3 years who are newly diagnosed with medulloblastoma and have metastatic disease or have had a subtotal resection include the following:
-
Surgery.
-
Adjuvant radiation therapy.
-
Adjuvant chemotherapy.
In high-risk patients, numerous studies have demonstrated that multimodality therapy improves the duration of disease control and overall disease-free survival.[58,134] Studies show that 50% to 70% of patients with high-risk disease, including those with metastatic disease, will experience long-term disease control.[58,117,134,135,136,137,138]; [139,140][Level of evidence A1]; [141][Level of evidence B4] A completed COG study demonstrated that children with group 3 MYC-amplified tumors who were randomly assigned to receive carboplatin during radiation therapy had improved 5-year EFS and OS rates, compared with those who did not receive carboplatin concurrently with radiation therapy.[135] The optimal treatment for children with SHH-activated, TP53-altered medulloblastoma has not been determined, as less than 30% of patients are expected to survive 5 years from diagnosis after treatment with current therapy.[14]
Surgery
Treatment of high-risk patients is the same as for average-risk patients. An attempt at gross-total resection is considered optimal, if feasible.[80,84]
Adjuvant radiation therapy
In contrast to standard-risk treatment, the craniospinal radiation dose is generally 36 Gy.
Adjuvant chemotherapy
Evidence (adjuvant chemotherapy):
- The drugs that are useful in children with average-risk disease are the same drugs that have been used extensively in children with high-risk disease, including cisplatin, lomustine, cyclophosphamide, etoposide, and vincristine.[139]
- These therapies have increased 5-year and 10-year EFS and OS rates and have likely decreased the incidence of late relapse.
- However, long-term survivors treated with multimodality therapy are at a high risk of late effects such as hearing loss, cardiac complications, and secondary neoplasms.[133]
- Postradiation high-dose nonmyeloablative chemotherapy supported by peripheral stem cell rescue, but with reduced cumulative doses of vincristine and cisplatin, has also been used and has resulted in 5-year PFS rates of approximately 60%.[58]
- The COG ACNS0332 (NCT00392327) study randomly assigned patients to receive daily carboplatin compared with weekly vincristine during radiation therapy (36 Gy craniospinal plus a posterior fossa boost) followed by six cycles of adjuvant treatment with cisplatin, cyclophosphamide, and vincristine.[135]
- Of the 261 evaluable patients, the 5-year EFS rate was 62.9%, and the OS rate was 73.4%.
- For the entire cohort, there was no difference in EFS rate between patients who were treated with carboplatin (66.4%) and patients who were not treated with carboplatin (59.2%).
- In a subset analysis based on molecular subgrouping, patients with group 3 medulloblastoma appeared to benefit from the use of daily carboplatin during radiation therapy, with a 5-year EFS rate of 73.2% for patients who received carboplatin, compared with 53.7% for those who did not.
- A second randomization testing the utility of isotretinoin maintenance therapy was closed at the time of a planned interim analysis for futility.
- In a trial of 51 patients with newly diagnosed high-risk disease, patients were treated with postoperative induction chemotherapy (etoposide and carboplatin), followed by two high-dose thiotepa courses with peripheral stem cell rescue and risk-adapted craniospinal radiation therapy.[136]
- The 5-year PFS and OS rates were 76%.
Treatment options under clinical evaluation for childhood medulloblastoma
Early-phase therapeutic trials may be available for selected patients. These trials may be available via the COG, the Pediatric Brain Tumor Consortium, or other entities. Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
References:
-
Zhang J, Walsh MF, Wu G, et al.: Germline Mutations in Predisposition Genes in Pediatric Cancer. N Engl J Med 373 (24): 2336-46, 2015.
-
Waszak SM, Northcott PA, Buchhalter I, et al.: Spectrum and prevalence of genetic predisposition in medulloblastoma: a retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol 19 (6): 785-798, 2018.
-
Hamilton SR, Liu B, Parsons RE, et al.: The molecular basis of Turcot's syndrome. N Engl J Med 332 (13): 839-47, 1995.
-
Taylor MD, Mainprize TG, Rutka JT, et al.: Medulloblastoma in a child with Rubenstein-Taybi Syndrome: case report and review of the literature. Pediatr Neurosurg 35 (5): 235-8, 2001.
-
Miller RW, Rubinstein JH: Tumors in Rubinstein-Taybi syndrome. Am J Med Genet 56 (1): 112-5, 1995.
-
Bourdeaut F, Miquel C, Richer W, et al.: Rubinstein-Taybi syndrome predisposing to non-WNT, non-SHH, group 3 medulloblastoma. Pediatr Blood Cancer 61 (2): 383-6, 2014.
-
Garrè ML, Cama A, Bagnasco F, et al.: Medulloblastoma variants: age-dependent occurrence and relation to Gorlin syndrome--a new clinical perspective. Clin Cancer Res 15 (7): 2463-71, 2009.
-
Pastorino L, Ghiorzo P, Nasti S, et al.: Identification of a SUFU germline mutation in a family with Gorlin syndrome. Am J Med Genet A 149A (7): 1539-43, 2009.
-
Brugières L, Remenieras A, Pierron G, et al.: High frequency of germline SUFU mutations in children with desmoplastic/nodular medulloblastoma younger than 3 years of age. J Clin Oncol 30 (17): 2087-93, 2012.
-
Evans DG, Farndon PA, Burnell LD, et al.: The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br J Cancer 64 (5): 959-61, 1991.
-
Smith MJ, Beetz C, Williams SG, et al.: Germline mutations in SUFU cause Gorlin syndrome-associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J Clin Oncol 32 (36): 4155-61, 2014.
-
Li FP, Fraumeni JF: Rhabdomyosarcoma in children: epidemiologic study and identification of a familial cancer syndrome. J Natl Cancer Inst 43 (6): 1365-73, 1969.
-
Pearson AD, Craft AW, Ratcliffe JM, et al.: Two families with the Li-Fraumeni cancer family syndrome. J Med Genet 19 (5): 362-5, 1982.
-
Kolodziejczak AS, Guerrini-Rousseau L, Planchon JM, et al.: Clinical outcome of pediatric medulloblastoma patients with Li-Fraumeni syndrome. Neuro Oncol 25 (12): 2273-2286, 2023.
-
Offit K, Levran O, Mullaney B, et al.: Shared genetic susceptibility to breast cancer, brain tumors, and Fanconi anemia. J Natl Cancer Inst 95 (20): 1548-51, 2003.
-
Dewire MD, Ellison DW, Patay Z, et al.: Fanconi anemia and biallelic BRCA2 mutation diagnosed in a young child with an embryonal CNS tumor. Pediatr Blood Cancer 53 (6): 1140-2, 2009.
-
Reid S, Renwick A, Seal S, et al.: Biallelic BRCA2 mutations are associated with multiple malignancies in childhood including familial Wilms tumour. J Med Genet 42 (2): 147-51, 2005.
-
Sönksen M, Obrecht-Sturm D, Hernáiz Driever P, et al.: Medulloblastoma in children with Fanconi anemia: Association with FA-D1/FA-N, SHH type and poor survival independent of treatment strategies. Neuro Oncol 26 (11): 2125-2139, 2024.
-
Begemann M, Waszak SM, Robinson GW, et al.: Germline GPR161 Mutations Predispose to Pediatric Medulloblastoma. J Clin Oncol 38 (1): 43-50, 2020.
-
Waszak SM, Robinson GW, Gudenas BL, et al.: Germline Elongator mutations in Sonic Hedgehog medulloblastoma. Nature 580 (7803): 396-401, 2020.
-
Rorke LB: The cerebellar medulloblastoma and its relationship to primitive neuroectodermal tumors. J Neuropathol Exp Neurol 42 (1): 1-15, 1983.
-
Louis DN, Perry A, Wesseling P, et al.: The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 23 (8): 1231-1251, 2021.
-
Ramaswamy V, Remke M, Shih D, et al.: Duration of the pre-diagnostic interval in medulloblastoma is subgroup dependent. Pediatr Blood Cancer 61 (7): 1190-4, 2014.
-
WHO Classification of Tumours Editorial Board, ed.: WHO Classification of Tumours: Central Nervous System Tumours. Vol. 6. 5th ed. IARC Press; 2021.
-
McManamy CS, Lamont JM, Taylor RE, et al.: Morphophenotypic variation predicts clinical behavior in childhood non-desmoplastic medulloblastomas. J Neuropathol Exp Neurol 62 (6): 627-32, 2003.
-
Eberhart CG, Kratz J, Wang Y, et al.: Histopathological and molecular prognostic markers in medulloblastoma: c-myc, N-myc, TrkC, and anaplasia. J Neuropathol Exp Neurol 63 (5): 441-9, 2004.
-
Giangaspero F, Perilongo G, Fondelli MP, et al.: Medulloblastoma with extensive nodularity: a variant with favorable prognosis. J Neurosurg 91 (6): 971-7, 1999.
-
Lafay-Cousin L, Bouffet E, Strother D, et al.: Phase II Study of Nonmetastatic Desmoplastic Medulloblastoma in Children Younger Than 4 Years of Age: A Report of the Children's Oncology Group (ACNS1221). J Clin Oncol 38 (3): 223-231, 2020.
-
Korshunov A, Okonechnikov K, Sahm F, et al.: Transcriptional profiling of medulloblastoma with extensive nodularity (MBEN) reveals two clinically relevant tumor subsets with VSNL1 as potent prognostic marker. Acta Neuropathol 139 (3): 583-596, 2020.
-
Onvani S, Etame AB, Smith CA, et al.: Genetics of medulloblastoma: clues for novel therapies. Expert Rev Neurother 10 (5): 811-23, 2010.
-
Dubuc AM, Northcott PA, Mack S, et al.: The genetics of pediatric brain tumors. Curr Neurol Neurosci Rep 10 (3): 215-23, 2010.
-
Thompson MC, Fuller C, Hogg TL, et al.: Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol 24 (12): 1924-31, 2006.
-
Kool M, Koster J, Bunt J, et al.: Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One 3 (8): e3088, 2008.
-
Tabori U, Baskin B, Shago M, et al.: Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J Clin Oncol 28 (8): 1345-50, 2010.
-
Pfister S, Remke M, Benner A, et al.: Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci. J Clin Oncol 27 (10): 1627-36, 2009.
-
Ellison DW, Onilude OE, Lindsey JC, et al.: beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children's Cancer Study Group Brain Tumour Committee. J Clin Oncol 23 (31): 7951-7, 2005.
-
Polkinghorn WR, Tarbell NJ: Medulloblastoma: tumorigenesis, current clinical paradigm, and efforts to improve risk stratification. Nat Clin Pract Oncol 4 (5): 295-304, 2007.
-
Giangaspero F, Wellek S, Masuoka J, et al.: Stratification of medulloblastoma on the basis of histopathological grading. Acta Neuropathol 112 (1): 5-12, 2006.
-
Northcott PA, Korshunov A, Witt H, et al.: Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29 (11): 1408-14, 2011.
-
Pomeroy SL, Tamayo P, Gaasenbeek M, et al.: Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature 415 (6870): 436-42, 2002.
-
Jones DT, Jäger N, Kool M, et al.: Dissecting the genomic complexity underlying medulloblastoma. Nature 488 (7409): 100-5, 2012.
-
Peyrl A, Chocholous M, Kieran MW, et al.: Antiangiogenic metronomic therapy for children with recurrent embryonal brain tumors. Pediatr Blood Cancer 59 (3): 511-7, 2012.
-
Taylor MD, Northcott PA, Korshunov A, et al.: Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123 (4): 465-72, 2012.
-
Kool M, Korshunov A, Remke M, et al.: Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 123 (4): 473-84, 2012.
-
Pietsch T, Schmidt R, Remke M, et al.: Prognostic significance of clinical, histopathological, and molecular characteristics of medulloblastomas in the prospective HIT2000 multicenter clinical trial cohort. Acta Neuropathol 128 (1): 137-49, 2014.
-
Cho YJ, Tsherniak A, Tamayo P, et al.: Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol 29 (11): 1424-30, 2011.
-
Gajjar A, Bowers DC, Karajannis MA, et al.: Pediatric Brain Tumors: Innovative Genomic Information Is Transforming the Diagnostic and Clinical Landscape. J Clin Oncol 33 (27): 2986-98, 2015.
-
Morrissy AS, Cavalli FMG, Remke M, et al.: Spatial heterogeneity in medulloblastoma. Nat Genet 49 (5): 780-788, 2017.
-
Wang X, Dubuc AM, Ramaswamy V, et al.: Medulloblastoma subgroups remain stable across primary and metastatic compartments. Acta Neuropathol 129 (3): 449-57, 2015.
-
Cavalli FMG, Remke M, Rampasek L, et al.: Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 31 (6): 737-754.e6, 2017.
-
Northcott PA, Buchhalter I, Morrissy AS, et al.: The whole-genome landscape of medulloblastoma subtypes. Nature 547 (7663): 311-317, 2017.
-
Schwalbe EC, Lindsey JC, Nakjang S, et al.: Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. Lancet Oncol 18 (7): 958-971, 2017.
-
Hicks D, Rafiee G, Schwalbe EC, et al.: The molecular landscape and associated clinical experience in infant medulloblastoma: prognostic significance of second-generation subtypes. Neuropathol Appl Neurobiol 47 (2): 236-250, 2021.
-
Northcott PA, Jones DT, Kool M, et al.: Medulloblastomics: the end of the beginning. Nat Rev Cancer 12 (12): 818-34, 2012.
-
Korshunov A, Sahm F, Zheludkova O, et al.: DNA methylation profiling is a method of choice for molecular verification of pediatric WNT-activated medulloblastomas. Neuro Oncol 21 (2): 214-221, 2019.
-
Gibson P, Tong Y, Robinson G, et al.: Subtypes of medulloblastoma have distinct developmental origins. Nature 468 (7327): 1095-9, 2010.
-
Ellison DW, Dalton J, Kocak M, et al.: Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol 121 (3): 381-96, 2011.
-
Gajjar A, Chintagumpala M, Ashley D, et al.: Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol 7 (10): 813-20, 2006.
-
Michalski JM, Janss AJ, Vezina LG, et al.: Children's Oncology Group Phase III Trial of Reduced-Dose and Reduced-Volume Radiotherapy With Chemotherapy for Newly Diagnosed Average-Risk Medulloblastoma. J Clin Oncol 39 (24): 2685-2697, 2021.
-
Goschzik T, Mynarek M, Doerner E, et al.: Genetic alterations of TP53 and OTX2 indicate increased risk of relapse in WNT medulloblastomas. Acta Neuropathol 144 (6): 1143-1156, 2022.
-
Gottardo NG: Genetic alterations of TP53 and OTX2 indicate increased risk of relapse in WNT medulloblastomas: "it's a numbers game"-implications for WNT medulloblastoma dose-reduction clinical trials. Acta Neuropathol 145 (1): 145-147, 2023.
-
Shuai S, Suzuki H, Diaz-Navarro A, et al.: The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature 574 (7780): 712-716, 2019.
-
Suzuki H, Kumar SA, Shuai S, et al.: Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature 574 (7780): 707-711, 2019.
-
Kool M, Jones DT, Jäger N, et al.: Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 25 (3): 393-405, 2014.
-
Robinson GW, Rudneva VA, Buchhalter I, et al.: Risk-adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol 19 (6): 768-784, 2018.
-
Leary SE, Zhou T, Holmes E, et al.: Histology predicts a favorable outcome in young children with desmoplastic medulloblastoma: a report from the children's oncology group. Cancer 117 (14): 3262-7, 2011.
-
Rutkowski S, von Hoff K, Emser A, et al.: Survival and prognostic factors of early childhood medulloblastoma: an international meta-analysis. J Clin Oncol 28 (33): 4961-8, 2010.
-
von Bueren AO, von Hoff K, Pietsch T, et al.: Treatment of young children with localized medulloblastoma by chemotherapy alone: results of the prospective, multicenter trial HIT 2000 confirming the prognostic impact of histology. Neuro Oncol 13 (6): 669-79, 2011.
-
Shih DJ, Northcott PA, Remke M, et al.: Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol 32 (9): 886-96, 2014.
-
Schwalbe EC, Williamson D, Lindsey JC, et al.: DNA methylation profiling of medulloblastoma allows robust subclassification and improved outcome prediction using formalin-fixed biopsies. Acta Neuropathol 125 (3): 359-71, 2013.
-
Zhukova N, Ramaswamy V, Remke M, et al.: Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol 31 (23): 2927-35, 2013.
-
Gajjar A, Robinson GW, Smith KS, et al.: Outcomes by Clinical and Molecular Features in Children With Medulloblastoma Treated With Risk-Adapted Therapy: Results of an International Phase III Trial (SJMB03). J Clin Oncol 39 (7): 822-835, 2021.
-
Goschzik T, Schwalbe EC, Hicks D, et al.: Prognostic effect of whole chromosomal aberration signatures in standard-risk, non-WNT/non-SHH medulloblastoma: a retrospective, molecular analysis of the HIT-SIOP PNET 4 trial. Lancet Oncol 19 (12): 1602-1616, 2018.
-
Gottardo NG, Hansford JR, McGlade JP, et al.: Medulloblastoma Down Under 2013: a report from the third annual meeting of the International Medulloblastoma Working Group. Acta Neuropathol 127 (2): 189-201, 2014.
-
Louis DN, Perry A, Burger P, et al.: International Society Of Neuropathology--Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol 24 (5): 429-35, 2014.
-
Sharma T, Schwalbe EC, Williamson D, et al.: Second-generation molecular subgrouping of medulloblastoma: an international meta-analysis of Group 3 and Group 4 subtypes. Acta Neuropathol 138 (2): 309-326, 2019.
-
Fouladi M, Gajjar A, Boyett JM, et al.: Comparison of CSF cytology and spinal magnetic resonance imaging in the detection of leptomeningeal disease in pediatric medulloblastoma or primitive neuroectodermal tumor. J Clin Oncol 17 (10): 3234-7, 1999.
-
Zeltzer PM, Boyett JM, Finlay JL, et al.: Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the Children's Cancer Group 921 randomized phase III study. J Clin Oncol 17 (3): 832-45, 1999.
-
Yao MS, Mehta MP, Boyett JM, et al.: The effect of M-stage on patterns of failure in posterior fossa primitive neuroectodermal tumors treated on CCG-921: a phase III study in a high-risk patient population. Int J Radiat Oncol Biol Phys 38 (3): 469-76, 1997.
-
Taylor RE, Bailey CC, Robinson K, et al.: Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma: The International Society of Paediatric Oncology/United Kingdom Children's Cancer Study Group PNET-3 Study. J Clin Oncol 21 (8): 1581-91, 2003.
-
Rutkowski S, Bode U, Deinlein F, et al.: Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med 352 (10): 978-86, 2005.
-
Rutkowski S, Gerber NU, von Hoff K, et al.: Treatment of early childhood medulloblastoma by postoperative chemotherapy and deferred radiotherapy. Neuro Oncol 11 (2): 201-10, 2009.
-
Ramaswamy V, Remke M, Adamski J, et al.: Medulloblastoma subgroup-specific outcomes in irradiated children: who are the true high-risk patients? Neuro Oncol 18 (2): 291-7, 2016.
-
Albright AL, Wisoff JH, Zeltzer PM, et al.: Effects of medulloblastoma resections on outcome in children: a report from the Children's Cancer Group. Neurosurgery 38 (2): 265-71, 1996.
-
Thompson EM, Hielscher T, Bouffet E, et al.: Prognostic value of medulloblastoma extent of resection after accounting for molecular subgroup: a retrospective integrated clinical and molecular analysis. Lancet Oncol 17 (4): 484-95, 2016.
-
Stargatt R, Rosenfeld JV, Maixner W, et al.: Multiple factors contribute to neuropsychological outcome in children with posterior fossa tumors. Dev Neuropsychol 32 (2): 729-48, 2007.
-
Pollack IF, Polinko P, Albright AL, et al.: Mutism and pseudobulbar symptoms after resection of posterior fossa tumors in children: incidence and pathophysiology. Neurosurgery 37 (5): 885-93, 1995.
-
Robertson PL, Muraszko KM, Holmes EJ, et al.: Incidence and severity of postoperative cerebellar mutism syndrome in children with medulloblastoma: a prospective study by the Children's Oncology Group. J Neurosurg 105 (6): 444-51, 2006.
-
Wells EM, Khademian ZP, Walsh KS, et al.: Postoperative cerebellar mutism syndrome following treatment of medulloblastoma: neuroradiographic features and origin. J Neurosurg Pediatr 5 (4): 329-34, 2010.
-
Gudrunardottir T, Sehested A, Juhler M, et al.: Cerebellar mutism: review of the literature. Childs Nerv Syst 27 (3): 355-63, 2011.
-
Wolfe-Christensen C, Mullins LL, Scott JG, et al.: Persistent psychosocial problems in children who develop posterior fossa syndrome after medulloblastoma resection. Pediatr Blood Cancer 49 (5): 723-6, 2007.
-
Jabarkheel R, Amayiri N, Yecies D, et al.: Molecular correlates of cerebellar mutism syndrome in medulloblastoma. Neuro Oncol 22 (2): 290-297, 2020.
-
Khan RB, Patay Z, Klimo P, et al.: Clinical features, neurologic recovery, and risk factors of postoperative posterior fossa syndrome and delayed recovery: a prospective study. Neuro Oncol 23 (9): 1586-1596, 2021.
-
Packer RJ, Gajjar A, Vezina G, et al.: Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol 24 (25): 4202-8, 2006.
-
Yock TI, Yeap BY, Ebb DH, et al.: Long-term toxic effects of proton radiotherapy for paediatric medulloblastoma: a phase 2 single-arm study. Lancet Oncol 17 (3): 287-98, 2016.
-
Eaton BR, Esiashvili N, Kim S, et al.: Clinical Outcomes Among Children With Standard-Risk Medulloblastoma Treated With Proton and Photon Radiation Therapy: A Comparison of Disease Control and Overall Survival. Int J Radiat Oncol Biol Phys 94 (1): 133-8, 2016.
-
Paulino AC, Mahajan A, Ye R, et al.: Ototoxicity and cochlear sparing in children with medulloblastoma: Proton vs. photon radiotherapy. Radiother Oncol 128 (1): 128-132, 2018.
-
Ris MD, Packer R, Goldwein J, et al.: Intellectual outcome after reduced-dose radiation therapy plus adjuvant chemotherapy for medulloblastoma: a Children's Cancer Group study. J Clin Oncol 19 (15): 3470-6, 2001.
-
Packer RJ, Sutton LN, Atkins TE, et al.: A prospective study of cognitive function in children receiving whole-brain radiotherapy and chemotherapy: 2-year results. J Neurosurg 70 (5): 707-13, 1989.
-
Johnson DL, McCabe MA, Nicholson HS, et al.: Quality of long-term survival in young children with medulloblastoma. J Neurosurg 80 (6): 1004-10, 1994.
-
Walter AW, Mulhern RK, Gajjar A, et al.: Survival and neurodevelopmental outcome of young children with medulloblastoma at St Jude Children's Research Hospital. J Clin Oncol 17 (12): 3720-8, 1999.
-
Laughton SJ, Merchant TE, Sklar CA, et al.: Endocrine outcomes for children with embryonal brain tumors after risk-adapted craniospinal and conformal primary-site irradiation and high-dose chemotherapy with stem-cell rescue on the SJMB-96 trial. J Clin Oncol 26 (7): 1112-8, 2008.
-
Geyer JR, Sposto R, Jennings M, et al.: Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children's Cancer Group. J Clin Oncol 23 (30): 7621-31, 2005.
-
Chi SN, Gardner SL, Levy AS, et al.: Feasibility and response to induction chemotherapy intensified with high-dose methotrexate for young children with newly diagnosed high-risk disseminated medulloblastoma. J Clin Oncol 22 (24): 4881-7, 2004.
-
Grill J, Sainte-Rose C, Jouvet A, et al.: Treatment of medulloblastoma with postoperative chemotherapy alone: an SFOP prospective trial in young children. Lancet Oncol 6 (8): 573-80, 2005.
-
Cohen BH, Geyer JR, Miller DC, et al.: Pilot Study of Intensive Chemotherapy With Peripheral Hematopoietic Cell Support for Children Less Than 3 Years of Age With Malignant Brain Tumors, the CCG-99703 Phase I/II Study. A Report From the Children's Oncology Group. Pediatr Neurol 53 (1): 31-46, 2015.
-
Duffner PK, Horowitz ME, Krischer JP, et al.: Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med 328 (24): 1725-31, 1993.
-
Ater JL, van Eys J, Woo SY, et al.: MOPP chemotherapy without irradiation as primary postsurgical therapy for brain tumors in infants and young children. J Neurooncol 32 (3): 243-52, 1997.
-
Grundy RG, Wilne SH, Robinson KJ, et al.: Primary postoperative chemotherapy without radiotherapy for treatment of brain tumours other than ependymoma in children under 3 years: results of the first UKCCSG/SIOP CNS 9204 trial. Eur J Cancer 46 (1): 120-33, 2010.
-
Blaney SM, Kocak M, Gajjar A, et al.: Pilot study of systemic and intrathecal mafosfamide followed by conformal radiation for infants with intracranial central nervous system tumors: a pediatric brain tumor consortium study (PBTC-001). J Neurooncol 109 (3): 565-71, 2012.
-
Mynarek M, von Hoff K, Pietsch T, et al.: Nonmetastatic Medulloblastoma of Early Childhood: Results From the Prospective Clinical Trial HIT-2000 and An Extended Validation Cohort. J Clin Oncol 38 (18): 2028-2040, 2020.
-
Dhall G, Grodman H, Ji L, et al.: Outcome of children less than three years old at diagnosis with non-metastatic medulloblastoma treated with chemotherapy on the "Head Start" I and II protocols. Pediatr Blood Cancer 50 (6): 1169-75, 2008.
-
Dhall G, O'Neil SH, Ji L, et al.: Excellent outcome of young children with nodular desmoplastic medulloblastoma treated on "Head Start" III: a multi-institutional, prospective clinical trial. Neuro Oncol 22 (12): 1862-1872, 2020.
-
Erker C, Mynarek M, Bailey S, et al.: Outcomes of Infants and Young Children With Relapsed Medulloblastoma After Initial Craniospinal Irradiation-Sparing Approaches: An International Cohort Study. J Clin Oncol 41 (10): 1921-1932, 2023.
-
Lafay-Cousin L, Smith A, Chi SN, et al.: Clinical, Pathological, and Molecular Characterization of Infant Medulloblastomas Treated with Sequential High-Dose Chemotherapy. Pediatr Blood Cancer 63 (9): 1527-34, 2016.
-
Ashley DM, Merchant TE, Strother D, et al.: Induction chemotherapy and conformal radiation therapy for very young children with nonmetastatic medulloblastoma: Children's Oncology Group study P9934. J Clin Oncol 30 (26): 3181-6, 2012.
-
Packer RJ, Sutton LN, Elterman R, et al.: Outcome for children with medulloblastoma treated with radiation and cisplatin, CCNU, and vincristine chemotherapy. J Neurosurg 81 (5): 690-8, 1994.
-
Bailey CC, Gnekow A, Wellek S, et al.: Prospective randomised trial of chemotherapy given before radiotherapy in childhood medulloblastoma. International Society of Paediatric Oncology (SIOP) and the (German) Society of Paediatric Oncology (GPO): SIOP II. Med Pediatr Oncol 25 (3): 166-78, 1995.
-
Kortmann RD, Kühl J, Timmermann B, et al.: Postoperative neoadjuvant chemotherapy before radiotherapy as compared to immediate radiotherapy followed by maintenance chemotherapy in the treatment of medulloblastoma in childhood: results of the German prospective randomized trial HIT '91. Int J Radiat Oncol Biol Phys 46 (2): 269-79, 2000.
-
Taylor RE, Bailey CC, Robinson KJ, et al.: Impact of radiotherapy parameters on outcome in the International Society of Paediatric Oncology/United Kingdom Children's Cancer Study Group PNET-3 study of preradiotherapy chemotherapy for M0-M1 medulloblastoma. Int J Radiat Oncol Biol Phys 58 (4): 1184-93, 2004.
-
von Hoff K, Hinkes B, Gerber NU, et al.: Long-term outcome and clinical prognostic factors in children with medulloblastoma treated in the prospective randomised multicentre trial HIT'91. Eur J Cancer 45 (7): 1209-17, 2009.
-
Cohen KJ, Munjapara V, Aguilera D, et al.: A Pilot Study Omitting Radiation in the Treatment of Children with Newly Diagnosed Wnt-Activated Medulloblastoma. Clin Cancer Res 29 (24): 5031-5037, 2023.
-
Thomas PR, Deutsch M, Kepner JL, et al.: Low-stage medulloblastoma: final analysis of trial comparing standard-dose with reduced-dose neuraxis irradiation. J Clin Oncol 18 (16): 3004-11, 2000.
-
Gupta T, Pervez S, Dasgupta A, et al.: Omission of Upfront Craniospinal Irradiation in Patients with Low-Risk WNT-Pathway Medulloblastoma Is Associated with Unacceptably High Risk of Neuraxial Failure. Clin Cancer Res 28 (19): 4180-4185, 2022.
-
Sabel M, Fleischhack G, Tippelt S, et al.: Relapse patterns and outcome after relapse in standard risk medulloblastoma: a report from the HIT-SIOP-PNET4 study. J Neurooncol 129 (3): 515-524, 2016.
-
Oyharcabal-Bourden V, Kalifa C, Gentet JC, et al.: Standard-risk medulloblastoma treated by adjuvant chemotherapy followed by reduced-dose craniospinal radiation therapy: a French Society of Pediatric Oncology Study. J Clin Oncol 23 (21): 4726-34, 2005.
-
Merchant TE, Kun LE, Krasin MJ, et al.: Multi-institution prospective trial of reduced-dose craniospinal irradiation (23.4 Gy) followed by conformal posterior fossa (36 Gy) and primary site irradiation (55.8 Gy) and dose-intensive chemotherapy for average-risk medulloblastoma. Int J Radiat Oncol Biol Phys 70 (3): 782-7, 2008.
-
Fukunaga-Johnson N, Sandler HM, Marsh R, et al.: The use of 3D conformal radiotherapy (3D CRT) to spare the cochlea in patients with medulloblastoma. Int J Radiat Oncol Biol Phys 41 (1): 77-82, 1998.
-
Huang E, Teh BS, Strother DR, et al.: Intensity-modulated radiation therapy for pediatric medulloblastoma: early report on the reduction of ototoxicity. Int J Radiat Oncol Biol Phys 52 (3): 599-605, 2002.
-
Carrie C, Grill J, Figarella-Branger D, et al.: Online quality control, hyperfractionated radiotherapy alone and reduced boost volume for standard risk medulloblastoma: long-term results of MSFOP 98. J Clin Oncol 27 (11): 1879-83, 2009.
-
Packer RJ, Goldwein J, Nicholson HS, et al.: Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy: A Children's Cancer Group Study. J Clin Oncol 17 (7): 2127-36, 1999.
-
Nageswara Rao AA, Wallace DJ, Billups C, et al.: Cumulative cisplatin dose is not associated with event-free or overall survival in children with newly diagnosed average-risk medulloblastoma treated with cisplatin based adjuvant chemotherapy: report from the Children's Oncology Group. Pediatr Blood Cancer 61 (1): 102-6, 2014.
-
Salloum R, Chen Y, Yasui Y, et al.: Late Morbidity and Mortality Among Medulloblastoma Survivors Diagnosed Across Three Decades: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 37 (9): 731-740, 2019.
-
Verlooy J, Mosseri V, Bracard S, et al.: Treatment of high risk medulloblastomas in children above the age of 3 years: a SFOP study. Eur J Cancer 42 (17): 3004-14, 2006.
-
Leary SES, Packer RJ, Li Y, et al.: Efficacy of Carboplatin and Isotretinoin in Children With High-risk Medulloblastoma: A Randomized Clinical Trial From the Children's Oncology Group. JAMA Oncol 7 (9): 1313-1321, 2021.
-
Dufour C, Foulon S, Geoffray A, et al.: Prognostic relevance of clinical and molecular risk factors in children with high-risk medulloblastoma treated in the phase II trial PNET HR+5. Neuro Oncol 23 (7): 1163-1172, 2021.
-
Gandola L, Massimino M, Cefalo G, et al.: Hyperfractionated accelerated radiotherapy in the Milan strategy for metastatic medulloblastoma. J Clin Oncol 27 (4): 566-71, 2009.
-
Evans AE, Jenkin RD, Sposto R, et al.: The treatment of medulloblastoma. Results of a prospective randomized trial of radiation therapy with and without CCNU, vincristine, and prednisone. J Neurosurg 72 (4): 572-82, 1990.
-
Jakacki RI, Burger PC, Zhou T, et al.: Outcome of children with metastatic medulloblastoma treated with carboplatin during craniospinal radiotherapy: a Children's Oncology Group Phase I/II study. J Clin Oncol 30 (21): 2648-53, 2012.
-
Tarbell NJ, Friedman H, Polkinghorn WR, et al.: High-risk medulloblastoma: a pediatric oncology group randomized trial of chemotherapy before or after radiation therapy (POG 9031). J Clin Oncol 31 (23): 2936-41, 2013.
-
von Bueren AO, Kortmann RD, von Hoff K, et al.: Treatment of Children and Adolescents With Metastatic Medulloblastoma and Prognostic Relevance of Clinical and Biologic Parameters. J Clin Oncol 34 (34): 4151-4160, 2016.
Childhood Nonmedulloblastoma Embryonal Tumors
The 2021 World Health Organization (WHO) Classification of Tumors of the Central Nervous System (CNS) is listed below in the Cellular and Molecular Classification section. All nonmedulloblastoma tumors of neuroectodermal origin that lack the specific histopathological features or molecular alterations that define other CNS tumors are classified as CNS embryonal tumors.[1,2] These tumors will be discussed in this section, with the exception of atypical teratoid/rhabdoid tumors (AT/RTs). For more information, see Childhood Central Nervous System Atypical Teratoid/Rhabdoid Tumor Treatment. Pineoblastoma will also be discussed in this summary because it shares histological features with the embryonal tumors and is conventionally treated in the same fashion. For more information, see the Childhood Pineoblastoma section.
Clinical Presentation
For nonmedulloblastoma embryonal tumors, presentation is also relatively rapid and depends on the location of the tumor in the nervous system. Embryonal tumors tend to grow fast and are usually diagnosed within 3 months of initial onset of symptoms.
Nonmedulloblastoma embryonal tumors may occur anywhere in the CNS, and presentation is variable. Usually there is significant neurological dysfunction associated with lethargy and vomiting. Supratentorial embryonal tumors (see Figure 1) result in focal neurological deficits such as hemiparesis and visual field loss, depending on which portion of the cerebral cortex is involved. They may also result in seizures and obtundation.
Cellular and Molecular Classification
The 2021 WHO Classification of Tumors of the CNS organizes nonmedulloblastoma embryonal tumors primarily by histological and immunohistological features and, in some cases, by molecular findings. The classification includes the following:[1,2]
- Atypical teratoid/rhabdoid tumor (AT/RT).
- Cribriform neuroepithelial tumor.
- Embryonal tumor with multilayered rosettes (ETMR).
- CNS neuroblastoma, FOXR2-activated.
- CNS tumor with BCOR internal tandem duplication.
- CNS embryonal tumor, not elsewhere classified (NEC)/not otherwise specified (NOS).
NEC is defined as a tumor not elsewhere classified. The NOS nomenclature is used for tumors that cannot be further classified. For many lesions, there are overlapping histological features, and methylation-based clustering is critical for specific diagnosis.[1,2] The contribution of DNA methylation profiling to correctly diagnose supratentorial embryonal tumors was demonstrated in a clinical trial of patients with supratentorial primitive neuroectodermal tumors of the CNS (CNS-PNET) and pineoblastoma.[3] For the pineoblastoma cases, there was high concordance between the diagnosis made by methylation profiling and the diagnosis made by central pathology review diagnosis (26 of 29). However, for the remaining 31 patients without pineoblastoma in the study, the diagnosis made by methylation profiling was high-grade glioma in 18 patients, AT/RT in 2 patients, and RELA fusion–positive ependymoma in 2 patients. Adjudication of discrepancies between the diagnosis made by central review pathology and the diagnosis made by methylation profiling was in favor of methylation profiling in the ten cases that were re-examined.
Subtypes of nonmedulloblastoma embryonal tumors
Molecular subtypes of nonmedulloblastoma embryonal tumors
Studies applying unsupervised clustering of DNA methylation patterns for nonmedulloblastoma embryonal tumors found that approximately one-half of these tumors diagnosed as nonmedulloblastoma embryonal tumors showed molecular profiles characteristic of other known pediatric brain tumors (e.g., high-grade gliomas).[4,5] These observations highlight the utility of molecular characterization to assign this class of tumors to their appropriate biology-based diagnosis.
Among the tumors diagnosed as nonmedulloblastoma embryonal tumors, molecular characterization identified genomically and biologically distinctive subtypes, including the following:
-
Cribriform neuroepithelial tumor: Representing less than 50% of nonmedulloblastoma embryonal tumors, this subtype is a nonrhabdoid tumor that arises in the vicinity of the third, fourth, or lateral ventricles. This tumor is characterized by cribriform strands and ribbons and demonstrates loss of nuclear SMARCB1 expression. The median age at diagnosis is 20 months. This tumor occurs more often in males, with a male-to-female ratio of 1.5 to 1.[6]
Genomic characterization of ten cases of cribriform neuroepithelial tumors showed large heterozygous 22q deletions in nine of ten cases with SMARCB1 variants on the alternative allele.[6] Cribriform neuroepithelial tumor showed DNA methylation profiles that matched those of the TYR subtype of atypical teratoid/rhabdoid tumor (AT/RT), a tumor that also arises in young children and shows loss of SMARCB1 expression. Patients with cribriform neuroepithelial tumors appear to have relatively favorable outcomes, in contrast to those of patients with AT/RT-TYR.[6]
-
Embryonal tumor with multilayered rosettes (ETMR): Representing up to 20% of nonmedulloblastoma embryonal tumors, this subtype combines embryonal, rosette-forming, neuroepithelial brain tumors that were previously categorized as either embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma, or medulloepithelioma.[4,7] ETMRs arise most commonly in young children (median age at diagnosis, 2–3 years) but may occur in older children.[5,7,8,9,10,11]
ETMRs are defined at the molecular level by high-level amplification of the microRNA cluster C19MC and by a gene fusion between TTYH1 and C19MC.[7,12,13] This gene fusion puts expression of C19MC under control of the TTYH1 promoter, leading to high-level aberrant expression of the microRNAs within the cluster. The World Health Organization (WHO) allows histologically similar tumors without C19MC alteration to be classified as ETMR, not otherwise specified (NOS). This subclass of tumors without C19MC alterations may harbor biallelic DICER1 variants.
-
Central nervous system (CNS) neuroblastoma with FOXR2 activation (CNS NB-FOXR2): Representing 10% to 15% of nonmedulloblastoma embryonal tumors, this tumor may occur in children younger than 3 years, but it more frequently occurs in older children. This subtype is characterized by genomic alterations that lead to increased expression of the transcription factor FOXR2.[4] CNS NB-FOXR2 is primarily observed in children younger than 10 years, and the histology of these tumors is typically that of CNS neuroblastoma or CNS ganglioneuroblastoma.[4,14] There is no single genomic alteration among CNS NB-FOXR2 tumors leading to FOXR2 overexpression, with gene fusions involving multiple FOXR2 partners identified.[4] Protein expression of SOX10 and ANKRD55 detected by immunohistochemistry has been proposed as a potential biomarker to differentiate CNS NB-FOXR2 tumors from related tumor types.[14]
-
CNS high-grade neuroepithelial tumor with BCOR alteration (CNS HGNET-BCOR): Representing up to 3% of nonmedulloblastoma embryonal tumors, this subtype is characterized by internal tandem duplications of BCOR,[4] a genomic alteration that is also found in clear cell sarcoma of the kidney.[15,16] While the median age at diagnosis is younger than 10 years, cases arising in the second decade of life and beyond do occur.[4]
Although not listed as separate entities in the 2021 WHO Classification of Tumors of the CNS, other nonmedulloblastoma embryonal tumors have also been described as separate entities, including the following:
-
CNS Ewing sarcoma family tumor with CIC alteration (CNS EFT-CIC): Representing 2% to 4% of nonmedulloblastoma embryonal tumors, this subtype is characterized by genomic alterations affecting CIC (located on chromosome 19q13.2), with fusion to NUTM1 being identified in several cases tested.[4,5]CIC gene fusions are also identified in extra-CNS Ewing-like sarcomas, and the gene expression signature of CNS EFT-CIC tumors is similar to that of these sarcomas.[4] CNS EFT-CIC tumors generally occur in children younger than 10 years and are characterized by a small cell phenotype but with variable histology.[4]
-
CNS high-grade neuroepithelial tumor with MN1 alteration (CNS HGNET-MN1): Representing 1% to 3% of nonmedulloblastoma embryonal tumors, this subtype is characterized by gene fusions involving MN1 (located on chromosome 22q12.3), with fusion partners including BEND2 and CXXC5.[4,5] The CNS HGNET-MN1 subtype shows a striking female predominance, and it tends to occur in the second decade of life.[4] This subtype contained most cases carrying a diagnosis of astroblastoma per the 2007 WHO classification scheme.[4] This subtype has not been added to the WHO diagnostic lexicon. Two other reports that together examined 35 cases of histologically defined astroblastoma found that 14 showed methylation profiles consistent with CNS HGNET-MN1 and/or MN1 alterations by fluorescence in situ hybridization.[17,18]
-
Medulloepithelioma: Medulloepithelioma with the classic C19MC amplification is considered an ETMR, C19MC-altered (see the ETMR information above). However, when a tumor has the histological features of medulloepithelioma, but without a C19MC amplification, it is still identified as an ETMR.[19,20] Medulloepithelioma tumors are rare and tend to arise most commonly in infants and young children. Medulloepitheliomas, which histologically recapitulate the embryonal neural tube, tend to arise supratentorially, primarily intraventricularly, but may arise infratentorially, in the cauda, and even extraneurally, along nerve roots.[19,20] Intraocular medulloepithelioma is biologically distinct from intra-axial medulloepithelioma.[21,22]
-
CNS embryonal tumor with PLAGL amplification: A retrospective analysis of more than 90,000 pediatric and adult brain tumors identified a small subset of embryonal tumors (n = 31) with distinct methylation profiles and high-level amplification and overexpression of either PLAGL1 or PLAGL2.[23] Additional recurrent genetic alterations observed in other pediatric CNS tumor types were not observed in these cases. These tumors occurred throughout the brain and were most commonly composed of primitive embryonal-like cells without markers of glial or neuronal differentiation. In this small cohort, differences in age at diagnosis and 10-year overall survival (OS) rates were reported between patients with PLAGL1 amplification (median age, 10.5 years; OS rate, 66%) and patients with PLAGL2 amplification (median age, 2 years; OS rate, 25%).
Staging Evaluation
Patients with nonmedulloblastoma embryonal tumors are staged in a fashion similar to that used for children with medulloblastoma. However, these patients are not assigned to average-risk and high-risk subgroups for treatment purposes because all patients are considered high risk. For more information, see the medulloblastoma Staging Evaluation section.
Treatment of Childhood Nonmedulloblastoma Embryonal Tumors
For more information about the treatment of CNS AT/RTs, see Childhood Central Nervous System Atypical Teratoid/Rhabdoid Tumor Treatment.
For more information about the treatment of medulloepithelioma, see the Treatment of Childhood Embryonal Tumors With Multilayered Rosettes or Medulloepithelioma section.
Treatment of children aged 3 years and younger
The optimal treatment of childhood nonmedulloblastoma embryonal tumors remains unclear and under study. Retrospective studies of fairly large numbers of patients have suggested management approaches for the more common subgroups, including AT/RTs, ETMRs, and FOXR2-activated tumors. For more information about the treatment of AT/RTs, see Childhood Central Nervous System Atypical Teratoid/Rhabdoid Tumor Treatment.
Standard treatment options for children aged 3 years and younger with newly diagnosed nonmedulloblastoma embryonal tumors, excluding AT/RTs, ETMRs, and FOXR2-activated tumors, include the following:
- Surgery.
- Adjuvant chemotherapy.
Treatment of children aged 3 years and younger with embryonal tumors is similar to that outlined for children aged 3 years and younger with medulloblastoma. Aggressive surgical resection is reasonable, given the improved rate of survival for medulloblastomas and other ETMRs after total or near-total resection.[11] For more information, see the Treatment of younger children with medulloblastoma section.
With the use of chemotherapy alone, outcome has been variable, with survival rates at 5 years ranging between 0% and 50%.[24,25,26]; [27][Level of evidence B4] The addition of craniospinal irradiation to chemotherapy-based regimens may successfully treat some children but with anticipated neurodevelopmental decline.[28][Level of evidence B4] Localized radiation therapy to the tumor site, either before or after chemotherapy, has been given, although data supporting its efficacy are unclear.
Treatment of children older than 3 years
Standard treatment options for children older than 3 years with newly diagnosed nonmedulloblastoma embryonal tumors, excluding AT/RTs, ETMRs, and FOXR2-activated tumors, include the following:
-
Surgery.
-
Adjuvant radiation therapy.
-
Adjuvant chemotherapy.
Surgery
Evidence (surgery):
- Nonmedulloblastoma embryonal tumors are often amenable to resection. In reported case series, 50% to 75% of tumors in patients were totally or near-totally resected.[29,30]; [3][Level of evidence A1]
- Attempting aggressive surgical resection is the first step in the management of newly diagnosed nonmedulloblastoma embryonal tumors. Although previous studies did not demonstrate that the extent of resection is predictive of outcome,[29,30,31] one study demonstrated improved survival when the tumor was completely resected.[32][Level of evidence B4] A published study (COG-ACNS0332 [NCT00392327]) of molecularly classified nonmedulloblastoma embryonal tumors revealed improved overall survival (OS) for patients who had less than 1.5 cm2 of residual disease, compared with patients who had more than 1.5 cm2 of residual disease.[3][Level of evidence A1]
Adjuvant radiation therapy
After surgery, children with nonmedulloblastoma embryonal tumors usually receive treatment similar to that received by children with high-risk medulloblastoma.
Conventionally, patients are treated with radiation to the entire neuraxis with local boost radiation therapy, as given for medulloblastoma.[31] Local boost radiation therapy may be problematic because of the size of the tumor and its location in the cerebral cortex. Also, there is no definitive evidence that craniospinal radiation therapy is superior to radiation to the primary tumor site alone in children with nondisseminated lesions.[29,30,31]
Adjuvant chemotherapy
The chemotherapeutic approaches during and after radiation therapy are similar to those used for children with high-risk medulloblastoma. The 3-year to 5-year OS rates range from 25% to 50%.[29,30,31]; [32,33][Level of evidence B4]; [34][Level of evidence C1]
In a published study of nonpineal tumors that were diagnosed as CNS primitive neuroectodermal tumors (PNETs) by traditional pathology, 71% of cases were revealed to be glioblastoma or another diagnosis by DNA methylation studies. Patients with nonmedulloblastoma embryonal tumors (n = 36) (including pineoblastomas, n = 26) had a 5-year OS rate of 78.5% (95% confidence interval [CI], 62.2%–94.8%). In contrast, the patients with glioblastoma had a 5-year OS rate of 12% (95% CI, 0%–24.7%). The study showed no benefit for children who received carboplatin or isotretinoin.[3][Level of evidence A1] This study highlights the importance of molecular classification of tumors traditionally termed CNS-PNET.[4]
Treatment of Childhood Embryonal Tumors With Multilayered Rosettes or Medulloepithelioma
A registry-based review of 159 patients with a confirmed molecular diagnosis of ETMR reported survival results for different treatments.[11]
- The 2-year event-free survival (EFS) and OS rates were 0% for patients treated with conventional chemotherapy without radiation therapy, regardless of the degree of surgical resection.
- The 2-year EFS rate was 21% and the OS rate was 30% for patients who had a gross-total resection and were treated with high-dose chemotherapy without radiation therapy.
- The 2-year EFS rate was 44% for children treated with high-dose chemotherapy and radiation therapy after a subtotal resection and 66% for patients treated with high-dose chemotherapy and radiation therapy after a gross-total resection.
- The relative roles of focal radiation therapy versus craniospinal radiation therapy could not be assessed in this review.
In a separate, but possibly overlapping, international retrospective review, 49 patients with histologically confirmed (by the treating institution) ETMRs were treated between 1988 to 2017 in a variety of ways.[35] The 5-year progression-free survival rate was 18% (± 6%), and the OS rate was 24% (± 6%). Most survivors received radiation therapy, including both local and craniospinal treatment, and there was no clear difference in outcomes between the types and extent of radiation therapy. The relative benefits of conventional chemotherapy compared with high-dose chemotherapy could not be assessed.[5]
In a subsequent publication, likely including some of the patients from the retrospective study, treatment was limited to only those who received chemotherapy and radiation therapy on the prospective P-HIT study or per the study protocol. The P-HIT study included postsurgery chemotherapy, high-dose chemotherapy, and radiation for some patients. In 35 patients with ETMRs, 8 long-term survivors were identified, 6 of whom had received either craniospinal or local radiation therapy, in addition to induction and high-dose chemotherapy. None of the patients who presented with brain stem disease survived. The 5-year survival was best for patients with localized disease, possibly for those treated with both induction and high-dose chemotherapy. The role of radiation therapy or the optimal volume of radiation therapy (local versus craniospinal) could not be determined.[10]
These studies suggest that the outcome for children with ETMRs may not be as dire as suggested by initial studies, which found a 5-year survival rate of 25% or lower. Outcome is more favorable in children with localized disease at the time of diagnosis and those who were treated with aggressive postsurgical chemotherapy, including induction and high-dose consolidation treatment. The role of radiation therapy is still unproven, and there is no evidence that craniospinal radiation in patients with localized disease is superior to focal radiation therapy.[5,10,11]
Treatment of CNS Neuroblastoma,FOXR2-Activated
The optimal treatment of patients with CNS neuroblastoma, FOXR2-activated tumors has not been confirmed by prospective studies. In a retrospective review of patients diagnosed between 1988 and 2007, the highest rates of survival were seen after complete or near-complete resections in patients with nonmetastatic tumors who also received craniospinal radiation therapy and possibly chemotherapy. With this type of approach, up to 75% of children (35 of 42) were alive 5 years after diagnosis and treatment. This tumor tends to occur in a somewhat older population than some of the other nonmedulloblastoma embryonal tumors.[5]
References:
-
Louis DN, Perry A, Wesseling P, et al.: The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 23 (8): 1231-1251, 2021.
-
WHO Classification of Tumours Editorial Board, ed.: WHO Classification of Tumours: Central Nervous System Tumours. Vol. 6. 5th ed. IARC Press; 2021.
-
Hwang EI, Kool M, Burger PC, et al.: Extensive Molecular and Clinical Heterogeneity in Patients With Histologically Diagnosed CNS-PNET Treated as a Single Entity: A Report From the Children's Oncology Group Randomized ACNS0332 Trial. J Clin Oncol : JCO2017764720, 2018.
-
Sturm D, Orr BA, Toprak UH, et al.: New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164 (5): 1060-72, 2016.
-
von Hoff K, Haberler C, Schmitt-Hoffner F, et al.: Therapeutic implications of improved molecular diagnostics for rare CNS embryonal tumor entities: results of an international, retrospective study. Neuro Oncol 23 (9): 1597-1611, 2021.
-
Johann PD, Hovestadt V, Thomas C, et al.: Cribriform neuroepithelial tumor: molecular characterization of a SMARCB1-deficient non-rhabdoid tumor with favorable long-term outcome. Brain Pathol 27 (4): 411-418, 2017.
-
Korshunov A, Sturm D, Ryzhova M, et al.: Embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma, and medulloepithelioma share molecular similarity and comprise a single clinicopathological entity. Acta Neuropathol 128 (2): 279-89, 2014.
-
Picard D, Miller S, Hawkins CE, et al.: Markers of survival and metastatic potential in childhood CNS primitive neuro-ectodermal brain tumours: an integrative genomic analysis. Lancet Oncol 13 (8): 838-48, 2012.
-
Spence T, Sin-Chan P, Picard D, et al.: CNS-PNETs with C19MC amplification and/or LIN28 expression comprise a distinct histogenetic diagnostic and therapeutic entity. Acta Neuropathol 128 (2): 291-303, 2014.
-
Juhnke BO, Gessi M, Gerber NU, et al.: Treatment of embryonal tumors with multilayered rosettes with carboplatin/etoposide induction and high-dose chemotherapy within the prospective P-HIT trial. Neuro Oncol 24 (1): 127-137, 2022.
-
Khan S, Solano-Paez P, Suwal T, et al.: Clinical phenotypes and prognostic features of embryonal tumours with multi-layered rosettes: a Rare Brain Tumor Registry study. Lancet Child Adolesc Health 5 (11): 800-813, 2021.
-
Kleinman CL, Gerges N, Papillon-Cavanagh S, et al.: Fusion of TTYH1 with the C19MC microRNA cluster drives expression of a brain-specific DNMT3B isoform in the embryonal brain tumor ETMR. Nat Genet 46 (1): 39-44, 2014.
-
Li M, Lee KF, Lu Y, et al.: Frequent amplification of a chr19q13.41 microRNA polycistron in aggressive primitive neuroectodermal brain tumors. Cancer Cell 16 (6): 533-46, 2009.
-
Korshunov A, Okonechnikov K, Schmitt-Hoffner F, et al.: Molecular analysis of pediatric CNS-PNET revealed nosologic heterogeneity and potent diagnostic markers for CNS neuroblastoma with FOXR2-activation. Acta Neuropathol Commun 9 (1): 20, 2021.
-
Ueno-Yokohata H, Okita H, Nakasato K, et al.: Consistent in-frame internal tandem duplications of BCOR characterize clear cell sarcoma of the kidney. Nat Genet 47 (8): 861-3, 2015.
-
Roy A, Kumar V, Zorman B, et al.: Recurrent internal tandem duplications of BCOR in clear cell sarcoma of the kidney. Nat Commun 6: 8891, 2015.
-
Wood MD, Tihan T, Perry A, et al.: Multimodal molecular analysis of astroblastoma enables reclassification of most cases into more specific molecular entities. Brain Pathol 28 (2): 192-202, 2018.
-
Lehman NL, Usubalieva A, Lin T, et al.: Genomic analysis demonstrates that histologically-defined astroblastomas are molecularly heterogeneous and that tumors with MN1 rearrangement exhibit the most favorable prognosis. Acta Neuropathol Commun 7 (1): 42, 2019.
-
Louis DN, Ohgaki H, Wiestler OD, et al.: The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114 (2): 97-109, 2007.
-
Sharma MC, Mahapatra AK, Gaikwad S, et al.: Pigmented medulloepithelioma: report of a case and review of the literature. Childs Nerv Syst 14 (1-2): 74-8, 1998 Jan-Feb.
-
Jakobiec FA, Kool M, Stagner AM, et al.: Intraocular Medulloepitheliomas and Embryonal Tumors With Multilayered Rosettes of the Brain: Comparative Roles of LIN28A and C19MC. Am J Ophthalmol 159 (6): 1065-1074.e1, 2015.
-
Korshunov A, Jakobiec FA, Eberhart CG, et al.: Comparative integrated molecular analysis of intraocular medulloepitheliomas and central nervous system embryonal tumors with multilayered rosettes confirms that they are distinct nosologic entities. Neuropathology 35 (6): 538-44, 2015.
-
Keck MK, Sill M, Wittmann A, et al.: Amplification of the PLAG-family genes-PLAGL1 and PLAGL2-is a key feature of the novel tumor type CNS embryonal tumor with PLAGL amplification. Acta Neuropathol 145 (1): 49-69, 2023.
-
Geyer JR, Sposto R, Jennings M, et al.: Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children's Cancer Group. J Clin Oncol 23 (30): 7621-31, 2005.
-
Marec-Berard P, Jouvet A, Thiesse P, et al.: Supratentorial embryonal tumors in children under 5 years of age: an SFOP study of treatment with postoperative chemotherapy alone. Med Pediatr Oncol 38 (2): 83-90, 2002.
-
Grill J, Sainte-Rose C, Jouvet A, et al.: Treatment of medulloblastoma with postoperative chemotherapy alone: an SFOP prospective trial in young children. Lancet Oncol 6 (8): 573-80, 2005.
-
Fangusaro J, Finlay J, Sposto R, et al.: Intensive chemotherapy followed by consolidative myeloablative chemotherapy with autologous hematopoietic cell rescue (AuHCR) in young children with newly diagnosed supratentorial primitive neuroectodermal tumors (sPNETs): report of the Head Start I and II experience. Pediatr Blood Cancer 50 (2): 312-8, 2008.
-
Friedrich C, von Bueren AO, von Hoff K, et al.: Treatment of young children with CNS-primitive neuroectodermal tumors/pineoblastomas in the prospective multicenter trial HIT 2000 using different chemotherapy regimens and radiotherapy. Neuro Oncol 15 (2): 224-34, 2013.
-
Cohen BH, Zeltzer PM, Boyett JM, et al.: Prognostic factors and treatment results for supratentorial primitive neuroectodermal tumors in children using radiation and chemotherapy: a Childrens Cancer Group randomized trial. J Clin Oncol 13 (7): 1687-96, 1995.
-
Reddy AT, Janss AJ, Phillips PC, et al.: Outcome for children with supratentorial primitive neuroectodermal tumors treated with surgery, radiation, and chemotherapy. Cancer 88 (9): 2189-93, 2000.
-
Timmermann B, Kortmann RD, Kühl J, et al.: Role of radiotherapy in the treatment of supratentorial primitive neuroectodermal tumors in childhood: results of the prospective German brain tumor trials HIT 88/89 and 91. J Clin Oncol 20 (3): 842-9, 2002.
-
Jakacki RI, Burger PC, Kocak M, et al.: Outcome and prognostic factors for children with supratentorial primitive neuroectodermal tumors treated with carboplatin during radiotherapy: a report from the Children's Oncology Group. Pediatr Blood Cancer 62 (5): 776-83, 2015.
-
Chintagumpala M, Hassall T, Palmer S, et al.: A pilot study of risk-adapted radiotherapy and chemotherapy in patients with supratentorial PNET. Neuro Oncol 11 (1): 33-40, 2009.
-
Johnston DL, Keene DL, Lafay-Cousin L, et al.: Supratentorial primitive neuroectodermal tumors: a Canadian pediatric brain tumor consortium report. J Neurooncol 86 (1): 101-8, 2008.
-
Liu APY, Dhanda SK, Lin T, et al.: Molecular classification and outcome of children with rare CNS embryonal tumors: results from St. Jude Children's Research Hospital including the multi-center SJYC07 and SJMB03 clinical trials. Acta Neuropathol 144 (4): 733-746, 2022.
Childhood Pineoblastoma
The World Health Organization classifies pineoblastomas in the tumors of the pineal region group. However, they are discussed in this summary because they share histological features with other embryonal tumors and are conventionally treated like other embryonal tumors.[1,2,3]
Clinical Presentation
Pineoblastoma often results in hydrocephalus due to blockage of cerebrospinal fluid at the third ventricular level and other symptoms related to pressure on the back of the brain stem in the tectal region. Symptoms may include a constellation of abnormalities in eye movements (Parinaud syndrome), manifested by pupils that react poorly to light but better to accommodation, loss of upgaze, retraction or convergence nystagmus, and lid retraction. As they grow, these tumors may also cause hemiparesis and ataxia.[4]
Cellular and Molecular Classification
Pineoblastoma is histologically similar to medulloblastoma and shares histological features with embryonal tumors. It is classified as a subgroup of pineal parenchymal tumors.[5,6]
Pineoblastoma can be classified into four distinctive subtypes with unique clinical and molecular characteristics:[7]
- The microRNA (miRNA) processing–altered 1 (PB-miRNA1) and miRNA processing–altered 2 (PB-miRNA2) subtypes are characterized by somatic or germline variants involving microRNA biogenesis genes (DICER1, DROSHA, and DGCR8). They are distinguished from each other by their DNA methylation profiles.
- The PB-MYC/FOXR2 subtype shows MYC activation and FOXR2 overexpression.
- The PB-RB1 subtype has RB1 alterations, and a minority of cases have a clinical diagnosis of trilateral retinoblastoma.
Additional information about each of the subtypes is provided below.
Genomics of Pineoblastoma
Pineoblastoma, which was previously conventionally grouped with embryonal tumors, is now categorized by the World Health Organization as a pineal parenchymal tumor. Given that therapies for pineoblastoma are quite similar to those used for embryonal tumors, the previous convention of including pineoblastoma with the central nervous system embryonal tumors is followed here. Pineoblastoma is associated with germline pathogenic variants in both the RB1 gene and the DICER1 gene, as described below:
- Pineoblastoma is associated with germline pathogenic variants in RB1. The term trilateral retinoblastoma is used to refer to ocular retinoblastoma in combination with a histologically similar brain tumor generally arising in the pineal gland or other midline structures. Historically, intracranial tumors have been reported in 5% to 15% of children with heritable retinoblastoma.[8] Rates of pineoblastoma among children with heritable retinoblastoma who undergo current treatment programs may be lower than these historical estimates.[9,10,11] In a study of patients with molecularly classified pineal parenchymal tumors, 6 of 221 cases (3%) had a clinical diagnosis of trilateral retinoblastoma.[7]
- Germline DICER1 pathogenic variants occur in some patients with pineoblastoma.[12] In one study, among 18 patients with pineoblastoma, 3 patients with DICER1 germline pathogenic variants were identified, and an additional 3 patients known to be carriers of germline DICER1 pathogenic variants developed pineoblastoma.[12] The DICER1 variants in patients with pineoblastoma are loss-of-function variants that appear to be distinct from the variants observed in DICER1 syndrome–related tumors such as pleuropulmonary blastoma.[12]
Genomic methods have been applied to pineoblastoma in an attempt to learn more about the tumor biology and guide future molecular classification. A retrospective, international meta-analysis included 221 children and adults diagnosed with pineoblastoma (n = 178) and pineal parenchymal tumors of intermediate differentiation (PPTID) (n = 43).[7] The evaluation identified four molecular groups of pineoblastoma based on DNA methylation, transcriptome profiling, and gene sequencing, as described below.
- The microRNA (miRNA) processing–altered 1 (PB-miRNA1) and miRNA processing–altered 2 (PB-miRNA2) subtypes are characterized by somatic or germline variants involving miRNA biogenesis genes (DICER1, DROSHA, and DGCR8).
- PB-miRNA1 represented approximately 50% of molecularly classified pineoblastoma cases, while PB-miRNA2 represented approximately 15% of the cases.
- The median age at presentation of PB-miRNA1 was approximately 8 years, and the median age at presentation of PB-miRNA2 was 12 years.
- The 5-year survival rate for patients with PB-miRNA2 (100%) exceeded that for patients with PB-miRNA1 (70%).
- The PB-MYC/FOXR2 subtype shows MYC activation (sometimes with MYC copy number gain and occasionally with MYC amplification) and FOXR2 overexpression.
- PB-MYC/FOXR2 represented approximately 20% of molecularly classified pineoblastoma cases.
- PB-MYC/FOXR2 cases presented at a young age (median, 1.4 years).
- Approximately 40% of patients with PB-MYC/FOXR2 presented with metastatic disease.
- The 5-year survival rate for patients with PB-MYC/FOXR2 was approximately 20%.
- The PB-RB1 subtype has RB1 alterations. In one study, 6 of 25 patients with the PB-RB1 subtype had a clinical diagnosis of trilateral retinoblastoma.
- The PB-RB1 subtype represented approximately 10% of molecularly classified pineoblastoma cases.
- Approximately 70% of PB-RB1 cases presented with metastatic disease.
- The 5-year survival rate for patients with PB-RB1 was approximately 30%.
- Cases with DNA methylation profiles indicating PPTID sometimes had a histological diagnosis of pineoblastoma, but the clinical and biological characteristics of these cases were distinctive from those of the pineoblastoma subtypes described above.
- Approximately 75% of cases with a molecular classification of PPTID had tumors with variants in KBTBD4, a gene that is also altered in group 3 and 4 medulloblastomas.
- Most PPTID cases occurred in adults, with a median age exceeding 30 years.
- The 5-year survival rate for patients with PPTID was 85%.
Staging Evaluation
Dissemination at the time of diagnosis occurs in 10% to 30% of patients with pineoblastoma.[13] Because of the location of the tumor, total resections are uncommon, and most patients have only a biopsy or a subtotal resection before postsurgical treatment.[13,14] Staging for children with pineoblastomas is the same as for children with medulloblastoma. However, the patients are not assigned to average-risk and high-risk subgroups for treatment purposes.[13] For more information, see the medulloblastoma Staging Evaluation section.
Treatment Option Overview for Childhood Pineoblastoma
Table 4 describes the treatment options for newly diagnosed and recurrent childhood pineoblastoma.
Treatment of Childhood Pineoblastoma
Treatment of children aged 3 years and younger
No standard treatment options currently exist for children aged 3 years and younger with pineoblastoma.[15] The following treatment approaches are available:
-
Biopsy (for diagnosis) and total resection, if possible.
-
Adjuvant chemotherapy.
-
High-dose, marrow-ablative chemotherapy with autologous bone marrow rescue or peripheral stem cell rescue.
Biopsy
Biopsy and, if possible, total resection, is usually performed to diagnose pineoblastoma.
Adjuvant chemotherapy
Children aged 3 years and younger with pineoblastoma are usually treated initially with chemotherapy in the hope of delaying, if not obviating, the need for radiation therapy.[16] Overall prognosis for this group remains very poor.[17,18,19] In two sequential, multicenter, prospective clinical trials, all five children younger than 3 years who were treated with chemotherapy died.[20][Level of evidence B4] In children responding to chemotherapy, the timing and amount of radiation therapy required after chemotherapy is unclear. The addition of craniospinal irradiation to chemotherapy-based regimens may successfully treat some children but with anticipated neurodevelopmental decline.[21][Level of evidence B4] Two large pooled analyses both revealed dismal survival for children younger than 3 or 4 years with pineoblastoma.[18,19]
High-dose, marrow-ablative chemotherapy with autologous bone marrow rescue or peripheral stem cell rescue
High-dose, marrow-ablative chemotherapy with autologous bone marrow rescue or peripheral stem cell rescue has been used with some success in young children.[22][Level of evidence B4] Two pooled analyses also revealed this modality may have some efficacy.[18,19]
Treatment of children older than 3 years
Standard treatment options for children older than 3 years with newly diagnosed pineoblastoma include the following:
-
Surgery.
-
Adjuvant radiation therapy.
-
Adjuvant chemotherapy.
Surgery
Surgery is usually the initial treatment for patients with pineoblastoma to diagnose the tumor.[23] Total resections have been associated with better outcomes.
Adjuvant radiation therapy
The usual postsurgical treatment for patients with pineoblastoma begins with radiation therapy, although some trials have used preradiation chemotherapy. The total dose of radiation therapy to the tumor site is 54 Gy to 55.8 Gy using conventional fractionation.[13,14]
Craniospinal irradiation with doses of 23.4 Gy to 36 Gy are also recommended because of the propensity of this tumor to disseminate throughout the subarachnoid space.[13,14,17]
Adjuvant chemotherapy
Chemotherapy is usually given in the same way as outlined for high-risk medulloblastomas in children with nondisseminated disease at the time of diagnosis.[15] For more information, see the Treatment of children older than 3 years with high-risk medulloblastoma section.
The 5-year disease-free survival rate exceeds 50% in children with localized disease at diagnosis who undergo aggressive resection.[13,14,24,25][Level of evidence A1] The Children's Oncology Group (COG) COG-ACNS0332 (NCT00392327) study of 36 patients with nonmedulloblastoma embryonal tumors (which included 26 pineoblastomas) reported a 5-year overall survival (OS) rate of 78.5% (95% confidence interval, 62.2%–94.8%).[25][Level of evidence A1]
For patients with disseminated disease at the time of diagnosis, survival is considerably poorer.[13,14] In the COG-ACNS0332 (NCT00392327) study, there was no significant difference in event-free survival or OS according to metastatic status.
Treatment options under clinical evaluation for childhood pineoblastoma
For patients with pineoblastoma, a variety of different treatment approaches are under evaluation, including the use of higher doses of chemotherapy after radiation therapy supported by peripheral stem cell rescue and the use of chemotherapy during radiation therapy.
Early-phase therapeutic trials may be available for selected patients. These trials may be available via the COG, the Pediatric Brain Tumor Consortium, or other entities. Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
References:
-
Louis DN, Perry A, Reifenberger G, et al.: The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131 (6): 803-20, 2016.
-
Louis DN, Perry A, Wesseling P, et al.: The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 23 (8): 1231-1251, 2021.
-
WHO Classification of Tumours Editorial Board, ed.: WHO Classification of Tumours: Central Nervous System Tumours. Vol. 6. 5th ed. IARC Press; 2021.
-
Chintagumpala MM, Paulino A, Panigrahy A, et al.: Embryonal and pineal region tumors. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Lippincott Williams and Wilkins, 2015, pp 671-99.
-
Li BK, Vasiljevic A, Dufour C, et al.: Pineoblastoma segregates into molecular sub-groups with distinct clinico-pathologic features: a Rare Brain Tumor Consortium registry study. Acta Neuropathol 139 (2): 223-241, 2020.
-
Pfaff E, Aichmüller C, Sill M, et al.: Molecular subgrouping of primary pineal parenchymal tumors reveals distinct subtypes correlated with clinical parameters and genetic alterations. Acta Neuropathol 139 (2): 243-257, 2020.
-
Liu APY, Li BK, Pfaff E, et al.: Clinical and molecular heterogeneity of pineal parenchymal tumors: a consensus study. Acta Neuropathol 141 (5): 771-785, 2021.
-
de Jong MC, Kors WA, de Graaf P, et al.: Trilateral retinoblastoma: a systematic review and meta-analysis. Lancet Oncol 15 (10): 1157-67, 2014.
-
Ramasubramanian A, Kytasty C, Meadows AT, et al.: Incidence of pineal gland cyst and pineoblastoma in children with retinoblastoma during the chemoreduction era. Am J Ophthalmol 156 (4): 825-9, 2013.
-
Abramson DH, Dunkel IJ, Marr BP, et al.: Incidence of pineal gland cyst and pineoblastoma in children with retinoblastoma during the chemoreduction era. Am J Ophthalmol 156 (6): 1319-20, 2013.
-
Turaka K, Shields CL, Meadows AT, et al.: Second malignant neoplasms following chemoreduction with carboplatin, etoposide, and vincristine in 245 patients with intraocular retinoblastoma. Pediatr Blood Cancer 59 (1): 121-5, 2012.
-
de Kock L, Sabbaghian N, Druker H, et al.: Germ-line and somatic DICER1 mutations in pineoblastoma. Acta Neuropathol 128 (4): 583-95, 2014.
-
Jakacki RI, Zeltzer PM, Boyett JM, et al.: Survival and prognostic factors following radiation and/or chemotherapy for primitive neuroectodermal tumors of the pineal region in infants and children: a report of the Childrens Cancer Group. J Clin Oncol 13 (6): 1377-83, 1995.
-
Timmermann B, Kortmann RD, Kühl J, et al.: Role of radiotherapy in the treatment of supratentorial primitive neuroectodermal tumors in childhood: results of the prospective German brain tumor trials HIT 88/89 and 91. J Clin Oncol 20 (3): 842-9, 2002.
-
Liu APY, Li BK, Vasiljevic A, et al.: SNO-EANO-EURACAN consensus on management of pineal parenchymal tumors. Neuro Oncol 26 (12): 2159-2173, 2024.
-
Mason WP, Grovas A, Halpern S, et al.: Intensive chemotherapy and bone marrow rescue for young children with newly diagnosed malignant brain tumors. J Clin Oncol 16 (1): 210-21, 1998.
-
Liu APY, Gudenas B, Lin T, et al.: Risk-adapted therapy and biological heterogeneity in pineoblastoma: integrated clinico-pathological analysis from the prospective, multi-center SJMB03 and SJYC07 trials. Acta Neuropathol 139 (2): 259-271, 2020.
-
Hansford JR, Huang J, Endersby R, et al.: Pediatric pineoblastoma: A pooled outcome study of North American and Australian therapeutic data. Neurooncol Adv 4 (1): vdac056, 2022.
-
Mynarek M, Pizer B, Dufour C, et al.: Evaluation of age-dependent treatment strategies for children and young adults with pineoblastoma: analysis of pooled European Society for Paediatric Oncology (SIOP-E) and US Head Start data. Neuro Oncol 19 (4): 576-585, 2017.
-
Hinkes BG, von Hoff K, Deinlein F, et al.: Childhood pineoblastoma: experiences from the prospective multicenter trials HIT-SKK87, HIT-SKK92 and HIT91. J Neurooncol 81 (2): 217-23, 2007.
-
Friedrich C, von Bueren AO, von Hoff K, et al.: Treatment of young children with CNS-primitive neuroectodermal tumors/pineoblastomas in the prospective multicenter trial HIT 2000 using different chemotherapy regimens and radiotherapy. Neuro Oncol 15 (2): 224-34, 2013.
-
Fangusaro J, Finlay J, Sposto R, et al.: Intensive chemotherapy followed by consolidative myeloablative chemotherapy with autologous hematopoietic cell rescue (AuHCR) in young children with newly diagnosed supratentorial primitive neuroectodermal tumors (sPNETs): report of the Head Start I and II experience. Pediatr Blood Cancer 50 (2): 312-8, 2008.
-
Jakacki RI, Burger PC, Kocak M, et al.: Outcome and prognostic factors for children with supratentorial primitive neuroectodermal tumors treated with carboplatin during radiotherapy: a report from the Children's Oncology Group. Pediatr Blood Cancer 62 (5): 776-83, 2015.
-
Gururangan S, McLaughlin C, Quinn J, et al.: High-dose chemotherapy with autologous stem-cell rescue in children and adults with newly diagnosed pineoblastomas. J Clin Oncol 21 (11): 2187-91, 2003.
-
Hwang EI, Kool M, Burger PC, et al.: Extensive Molecular and Clinical Heterogeneity in Patients With Histologically Diagnosed CNS-PNET Treated as a Single Entity: A Report From the Children's Oncology Group Randomized ACNS0332 Trial. J Clin Oncol : JCO2017764720, 2018.
Treatment of Recurrent Childhood Medulloblastoma and Other CNS Embryonal Tumors
Recurrence of all forms of central nervous system (CNS) embryonal tumors is not uncommon and usually occurs within 36 months of treatment. However, recurrent tumors may also develop many years after initial treatment.[1,2,3] In such late relapses, especially those occurring 5 or more years after diagnosis, differentiation from secondary tumors such as high-grade gliomas can be difficult. Histological confirmation is recommended and usually required. In a 2021 report, a paired molecular cohort was assembled, consisting of 127 patients with tissue specimens available from both their primary medulloblastomas and subsequent tumors associated with relapse. Comparative molecular analyses were performed using the patient-matched tumor specimens. DNA methylation-based classification identified nine relapse cases (7%) as histologies other than medulloblastoma.[4] Disease may recur at the primary site or may be disseminated at the time of relapse. Sites of noncontiguous relapse may include the spinal leptomeninges, intracranial sites, and cerebrospinal fluid, in isolation or in any combination, and may be associated with primary tumor relapse.[1,2,5] Extraneural disease relapse is rare and is seen primarily in patients who were treated with radiation therapy alone.[6][Level of evidence C1]
Studies have found that even in patients with nondisseminated disease at diagnosis, and independent of the dose of radiation therapy or the type of chemotherapy, approximately one-third of patients will experience a relapse at the primary tumor site alone, one-third at the primary tumor site plus distant sites, and one-third at distant sites without relapse at the primary site.[1,2,5]
Treatment Options
There are no standard treatment options for recurrent childhood CNS embryonal tumors. For more information, see the Treatment for Recurrent Childhood CNS Atypical Teratoid/Rhabdoid Tumor section in Childhood Central Nervous System Atypical Teratoid/Rhabdoid Tumor Treatment.
For most children, treatment is palliative, and disease control is transient in patients previously treated with radiation therapy and chemotherapy, with more than 80% of patients progressing within 2 years.[3]; [7][Level of evidence C1] The temporal and spatial patterns of relapse differ between molecular subgroups. Patients with group 4 medulloblastomas present with delayed relapse compared with patients in other subgroups. In children who develop relapsed disease after radiation-sparing strategies, patients with group 4 and SHH medulloblastomas have higher rates of local relapse (42% and 39%, respectively), compared with patients with group 3 disease (17%).[8] For young children, predominantly those younger than 3 years at diagnosis who were never treated with radiation therapy, longer-term control with reoperation, radiation therapy, and chemotherapy is possible.[5,8,9,10,11,12]
Treatment approaches may include the following:
-
Surgery.
-
Radiation therapy.
-
Chemotherapy.
-
High-dose chemotherapy with stem cell rescue.
-
Molecularly targeted therapy.
Surgery
At the time of relapse, a complete evaluation for extent of recurrence is indicated for all embryonal tumors. Biopsy or surgical resection may be necessary for confirmation of relapse because other entities, such as secondary tumors and treatment-related brain necrosis, may be clinically indistinguishable from tumor recurrence. The need for surgical intervention must be individualized based on the initial tumor type, the length of time between initial treatment and the reappearance of the lesion, and clinical symptoms.
Radiation therapy
Patients with recurrent embryonal tumors who have already received radiation therapy and chemotherapy may be candidates for further radiation therapy depending on the site and dose of previous radiation. Treatment may include reirradiation at the primary tumor site, focal areas of radiation therapy to sites of disseminated disease, and craniospinal irradiation (CSI).[13,14] However, long-term survival has been observed in a subset of patients who received chemotherapy alone at the time of diagnosis and had local relapse. This finding was primarily noted in young children with SHH-activated disease.[8,15] In most cases, such therapy is palliative. Stereotactic radiation therapy and/or salvage chemotherapy can also be used.[16] For more information, see the Chemotherapy section.
- One retrospective study reported the outcomes of infants and young children with relapsed medulloblastoma who were initially treated in a variety of different chemotherapy clinical trials without CSI.[8]
- At the time of relapse, 73% of these children were treated with CSI-based regimens.
- The 3-year postrelapse survival rate was 52.4% for patients treated with curative intent.
- The 3-year postrelapse survival rates for children with SHH, group 3, and group 4 medulloblastoma who received salvage radiation therapy were 61%, 40%, and 79%, respectively. Patients with SHH disease were less likely to receive salvage radiation therapy.
- Older age at diagnosis, local relapse, and the SHH infant subtype were associated with better postrelapse survival.
Chemotherapy
Recurrent CNS embryonal tumors can respond to chemotherapeutic agents used singularly or in combination, including cyclophosphamide, cisplatin, carboplatin, lomustine, etoposide, topotecan, temozolomide, the combination of irinotecan and temozolomide with or without bevacizumab, and antiangiogenic metronomic therapy.[9,17,18,19,20,21,22,23,24,25,26,27]; [28,29,30][Level of evidence B4] Approximately 30% to 50% of these patients have objective responses to conventional chemotherapy, but long-term disease control is rare.
For select patients with recurrent medulloblastoma—primarily infants and young children who were treated at the time of diagnosis with chemotherapy alone and who developed local recurrence—long-term disease control may be obtained after further treatment with chemotherapy plus local radiation therapy. This potential may be greatest in patients who are able to undergo complete resection of the recurrent disease.[31][Level of evidence B4]; [32][Level of evidence C1]
In a St. Jude Children's Research Hospital study (SJYC07 [NCT00602667]), 29 patients with progressive disease received CSI (median dose, 36 Gy; interquartile range, 36–36). Of these 29 patients, 18 (62%) were alive at the time of publication, compared with 6 of 25 patients (24%) who did not receive CSI.[12][Level of evidence B4]
High-dose chemotherapy with stem cell rescue
For patients who have previously received radiation therapy, higher-dose chemotherapeutic regimens, supported with autologous bone marrow rescue or peripheral stem cell support, have been used with variable results.[10,11,33,34,35,36][Level of evidence B4]; [37,38,39][Level of evidence C1]
- With such regimens, objective response is frequent, occurring in 50% to 75% of patients. However, long-term disease control is obtained in fewer than 30% of patients and is seen primarily in patients in first relapse and those with only localized disease at the time of relapse.[11]; [36][Level of evidence B4]; [37][Level of evidence C1]
- Additionally, results from national trials for relapsed medulloblastoma that specified intent to transplant as part of their treatment plan showed that only approximately 5% of patients initiating retrieval therapy achieved long-term disease-free survival with this strategy.[36,40] Thus, studies that report from the time of transplant overestimate the benefit of transplant-based approaches for the total population of patients who have a relapse.
- Long-term disease control for patients with disseminated disease is infrequent.[41][Level of evidence C1]
Molecularly targeted therapy
With increased knowledge of the molecular and genetic changes associated with different subtypes of medulloblastoma, molecularly targeted therapy, also called precision therapy, is being actively explored in children with recurrent disease.
In patients with recurrent SHH-activated medulloblastomas, the SHH PTCH1 inhibitor vismodegib demonstrated radiographic responses in 3 of 12 pediatric patients. Two of the responses were sustained for less than 2 months, and one response was sustained for more than 6 months. Response was only seen in patients with upstream variants of the SHH pathway, at the level of PTCH1 or SMO.[42] However, because of the development of irreversible growth plate fusions, the use of vismodegib is limited to skeletally mature children.[43]
Treatment Options Under Clinical Evaluation for Recurrent Childhood Medulloblastoma and Other CNS Embryonal Tumors
Early-phase therapeutic trials may be available for selected patients. These trials may be available via the Children's Oncology Group (COG), the Pediatric Brain Tumor Consortium, or other entities. Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
-
Taylor RE, Bailey CC, Robinson K, et al.: Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma: The International Society of Paediatric Oncology/United Kingdom Children's Cancer Study Group PNET-3 Study. J Clin Oncol 21 (8): 1581-91, 2003.
-
Packer RJ, Goldwein J, Nicholson HS, et al.: Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy: A Children's Cancer Group Study. J Clin Oncol 17 (7): 2127-36, 1999.
-
Sabel M, Fleischhack G, Tippelt S, et al.: Relapse patterns and outcome after relapse in standard risk medulloblastoma: a report from the HIT-SIOP-PNET4 study. J Neurooncol 129 (3): 515-524, 2016.
-
Kumar R, Smith KS, Deng M, et al.: Clinical Outcomes and Patient-Matched Molecular Composition of Relapsed Medulloblastoma. J Clin Oncol 39 (7): 807-821, 2021.
-
Oyharcabal-Bourden V, Kalifa C, Gentet JC, et al.: Standard-risk medulloblastoma treated by adjuvant chemotherapy followed by reduced-dose craniospinal radiation therapy: a French Society of Pediatric Oncology Study. J Clin Oncol 23 (21): 4726-34, 2005.
-
Mazloom A, Zangeneh AH, Paulino AC: Prognostic factors after extraneural metastasis of medulloblastoma. Int J Radiat Oncol Biol Phys 78 (1): 72-8, 2010.
-
Johnston DL, Keene D, Strother D, et al.: Survival Following Tumor Recurrence in Children With Medulloblastoma. J Pediatr Hematol Oncol 40 (3): e159-e163, 2018.
-
Erker C, Mynarek M, Bailey S, et al.: Outcomes of Infants and Young Children With Relapsed Medulloblastoma After Initial Craniospinal Irradiation-Sparing Approaches: An International Cohort Study. J Clin Oncol 41 (10): 1921-1932, 2023.
-
Gentet JC, Doz F, Bouffet E, et al.: Carboplatin and VP 16 in medulloblastoma: a phase II Study of the French Society of Pediatric Oncology (SFOP). Med Pediatr Oncol 23 (5): 422-7, 1994.
-
Kadota RP, Mahoney DH, Doyle J, et al.: Dose intensive melphalan and cyclophosphamide with autologous hematopoietic stem cells for recurrent medulloblastoma or germinoma. Pediatr Blood Cancer 51 (5): 675-8, 2008.
-
Butturini AM, Jacob M, Aguajo J, et al.: High-dose chemotherapy and autologous hematopoietic progenitor cell rescue in children with recurrent medulloblastoma and supratentorial primitive neuroectodermal tumors: the impact of prior radiotherapy on outcome. Cancer 115 (13): 2956-63, 2009.
-
Robinson GW, Rudneva VA, Buchhalter I, et al.: Risk-adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol 19 (6): 768-784, 2018.
-
Wetmore C, Herington D, Lin T, et al.: Reirradiation of recurrent medulloblastoma: does clinical benefit outweigh risk for toxicity? Cancer 120 (23): 3731-7, 2014.
-
Baroni LV, Freytes C, Fernández Ponce N, et al.: Craniospinal irradiation as part of re-irradiation for children with recurrent medulloblastoma. J Neurooncol 155 (1): 53-61, 2021.
-
Hill RM, Richardson S, Schwalbe EC, et al.: Time, pattern, and outcome of medulloblastoma relapse and their association with tumour biology at diagnosis and therapy: a multicentre cohort study. Lancet Child Adolesc Health 4 (12): 865-874, 2020.
-
Abe M, Tokumaru S, Tabuchi K, et al.: Stereotactic radiation therapy with chemotherapy in the management of recurrent medulloblastomas. Pediatr Neurosurg 42 (2): 81-8, 2006.
-
Friedman HS, Oakes WJ: The chemotherapy of posterior fossa tumors in childhood. J Neurooncol 5 (3): 217-29, 1987.
-
Needle MN, Molloy PT, Geyer JR, et al.: Phase II study of daily oral etoposide in children with recurrent brain tumors and other solid tumors. Med Pediatr Oncol 29 (1): 28-32, 1997.
-
Gaynon PS, Ettinger LJ, Baum ES, et al.: Carboplatin in childhood brain tumors. A Children's Cancer Study Group Phase II trial. Cancer 66 (12): 2465-9, 1990.
-
Allen JC, Walker R, Luks E, et al.: Carboplatin and recurrent childhood brain tumors. J Clin Oncol 5 (3): 459-63, 1987.
-
Ashley DM, Longee D, Tien R, et al.: Treatment of patients with pineoblastoma with high dose cyclophosphamide. Med Pediatr Oncol 26 (6): 387-92, 1996.
-
Lefkowitz IB, Packer RJ, Siegel KR, et al.: Results of treatment of children with recurrent medulloblastoma/primitive neuroectodermal tumors with lomustine, cisplatin, and vincristine. Cancer 65 (3): 412-7, 1990.
-
Friedman HS, Mahaley MS, Schold SC, et al.: Efficacy of vincristine and cyclophosphamide in the therapy of recurrent medulloblastoma. Neurosurgery 18 (3): 335-40, 1986.
-
Castello MA, Clerico A, Deb G, et al.: High-dose carboplatin in combination with etoposide (JET regimen) for childhood brain tumors. Am J Pediatr Hematol Oncol 12 (3): 297-300, 1990.
-
Cefalo G, Massimino M, Ruggiero A, et al.: Temozolomide is an active agent in children with recurrent medulloblastoma/primitive neuroectodermal tumor: an Italian multi-institutional phase II trial. Neuro Oncol 16 (5): 748-53, 2014.
-
Le Teuff G, Castaneda-Heredia A, Dufour C, et al.: Phase II study of temozolomide and topotecan (TOTEM) in children with relapsed or refractory extracranial and central nervous system tumors including medulloblastoma with post hoc Bayesian analysis: A European ITCC study. Pediatr Blood Cancer 67 (1): e28032, 2020.
-
Levy AS, Krailo M, Chi S, et al.: Temozolomide with irinotecan versus temozolomide, irinotecan plus bevacizumab for recurrent medulloblastoma of childhood: Report of a COG randomized Phase II screening trial. Pediatr Blood Cancer 68 (8): e29031, 2021.
-
Minturn JE, Janss AJ, Fisher PG, et al.: A phase II study of metronomic oral topotecan for recurrent childhood brain tumors. Pediatr Blood Cancer 56 (1): 39-44, 2011.
-
Peyrl A, Chocholous M, Kieran MW, et al.: Antiangiogenic metronomic therapy for children with recurrent embryonal brain tumors. Pediatr Blood Cancer 59 (3): 511-7, 2012.
-
Peyrl A, Chocholous M, Sabel M, et al.: Sustained Survival Benefit in Recurrent Medulloblastoma by a Metronomic Antiangiogenic Regimen: A Nonrandomized Controlled Trial. JAMA Oncol 9 (12): 1688-1695, 2023.
-
Ridola V, Grill J, Doz F, et al.: High-dose chemotherapy with autologous stem cell rescue followed by posterior fossa irradiation for local medulloblastoma recurrence or progression after conventional chemotherapy. Cancer 110 (1): 156-63, 2007.
-
Bakst RL, Dunkel IJ, Gilheeney S, et al.: Reirradiation for recurrent medulloblastoma. Cancer 117 (21): 4977-82, 2011.
-
Dunkel IJ, Boyett JM, Yates A, et al.: High-dose carboplatin, thiotepa, and etoposide with autologous stem-cell rescue for patients with recurrent medulloblastoma. Children's Cancer Group. J Clin Oncol 16 (1): 222-8, 1998.
-
Park JE, Kang J, Yoo KH, et al.: Efficacy of high-dose chemotherapy and autologous stem cell transplantation in patients with relapsed medulloblastoma: a report on the Korean Society for Pediatric Neuro-Oncology (KSPNO)-S-053 study. J Korean Med Sci 25 (8): 1160-6, 2010.
-
Gilman AL, Jacobsen C, Bunin N, et al.: Phase I study of tandem high-dose chemotherapy with autologous peripheral blood stem cell rescue for children with recurrent brain tumors: a Pediatric Blood and MarrowTransplant Consortium study. Pediatr Blood Cancer 57 (3): 506-13, 2011.
-
Pizer B, Donachie PH, Robinson K, et al.: Treatment of recurrent central nervous system primitive neuroectodermal tumours in children and adolescents: results of a Children's Cancer and Leukaemia Group study. Eur J Cancer 47 (9): 1389-97, 2011.
-
Massimino M, Gandola L, Spreafico F, et al.: No salvage using high-dose chemotherapy plus/minus reirradiation for relapsing previously irradiated medulloblastoma. Int J Radiat Oncol Biol Phys 73 (5): 1358-63, 2009.
-
Gururangan S, Krauser J, Watral MA, et al.: Efficacy of high-dose chemotherapy or standard salvage therapy in patients with recurrent medulloblastoma. Neuro Oncol 10 (5): 745-51, 2008.
-
Dunkel IJ, Gardner SL, Garvin JH, et al.: High-dose carboplatin, thiotepa, and etoposide with autologous stem cell rescue for patients with previously irradiated recurrent medulloblastoma. Neuro Oncol 12 (3): 297-303, 2010.
-
Gajjar A, Pizer B: Role of high-dose chemotherapy for recurrent medulloblastoma and other CNS primitive neuroectodermal tumors. Pediatr Blood Cancer 54 (4): 649-51, 2010.
-
Bowers DC, Gargan L, Weprin BE, et al.: Impact of site of tumor recurrence upon survival for children with recurrent or progressive medulloblastoma. J Neurosurg 107 (1 Suppl): 5-10, 2007.
-
Robinson GW, Orr BA, Wu G, et al.: Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J Clin Oncol 33 (24): 2646-54, 2015.
-
Robinson GW, Kaste SC, Chemaitilly W, et al.: Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget 8 (41): 69295-69302, 2017.
Latest Updates to This Summary (04 / 11 / 2025)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
This summary was comprehensively reviewed.
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of childhood medulloblastoma and other central nervous system embryonal tumors. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Childhood Medulloblastoma and Other Central Nervous System Embryonal Tumors Treatment are:
- Kenneth J. Cohen, MD, MBA (Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins Hospital)
- Louis S. Constine, MD (James P. Wilmot Cancer Center at University of Rochester Medical Center)
- Roger J. Packer, MD (Children's National Hospital)
- Malcolm A. Smith, MD, PhD (National Cancer Institute)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as "NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary]."
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Childhood Medulloblastoma and Other Central Nervous System Embryonal Tumors Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/brain/hp/child-cns-embryonal-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389418]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either "standard" or "under clinical evaluation." These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website's Email Us.
Last Revised: 2025-04-11

