Langerhans Cell Histiocytosis Treatment (PDQ®): Treatment - Health Professional Information [NCI]
This information is produced and provided by the National Cancer Institute (NCI). The information in this topic may have changed since it was written. For the most current information, contact the National Cancer Institute via the Internet web site at http://cancer.gov or call 1-800-4-CANCER.
General Information About Langerhans Cell Histiocytosis (LCH)
Histiocytic diseases in children and adults are caused by an abnormal accumulation of cells of the mononuclear phagocytic system. This summary discusses only Langerhans cell histiocytosis (LCH), a myeloid-derived dendritic cell disorder.
Histiocytic diseases have been reclassified into five categories, with LCH in the L group (see Table 1).[1,2] LCH results from the clonal proliferation of immunophenotypically and functionally immature, morphologically rounded LCH cells found in relevant lesions, along with eosinophils, macrophages, lymphocytes, and, occasionally, multinucleated giant cells.[3,4] The pathological histiocytes and normal Langerhans cells of the epidermis (LCs) have identical immunophenotypic characteristics, including the presence of Birbeck granules identified by electron microscopy. There are clear morphological, phenotypic, and gene expression differences between the pathological variant of the LCH lesions (LCH cells) and the normal LCs, hence the term LCH cells.
| Histiocytosis Group | Diseases | |
|---|---|---|
| AXG = adult xanthogranuloma; BCH = benign cephalic histiocytosis; GEH = generalized eruptive histiocytosis; HLH = hemophagocytic lymphohistiocytosis; JXG = juvenile xanthogranuloma; LCH = Langerhans cell histiocytosis; MRH = multicentric reticulohistiocytosis; NXG = necrobiotic xanthogranuloma; PNH = progressive nodular histiocytosis; RDD = Rosai-Dorfman disease; SRH = solitary reticulohistiocytoma; XD = xanthoma disseminatum. | ||
| a Adapted from Emile et al.[2] | ||
| b Reprinted fromBlood, Volume 135, Issue 16, Carlos Rodriguez-Galindo, Carl E. Allen, Langerhans cell histiocytosis, Pages 1319–1331, Copyright 2020, with permission from Elsevier.[1] | ||
| L Group | LCH | |
| Indeterminate-cell histiocytosis (ICH) | ||
| Erdheim-Chester disease (ECD) | ||
| Mixed LCH/ECD | ||
| C Group | Cutaneous non-LCH | |
| Xanthomatous granuloma (XG) family: JXG, AXG, SRH, BCH, GEH, PNH | ||
| Non-XG family: Cutaneous RDD, NXG, other | ||
| Cutaneous non-LCH with a major systemic component | ||
| XG family: XD | ||
| Non-XG family: MRH | ||
| R Group | Familial RDD | |
| Sporadic RDD | ||
| Classical RDD | ||
| Extranodal RDD | ||
| RDD with neoplasia or immune disease | ||
| Unclassified | ||
| M Group | Primary malignant histiocytoses | |
| Secondary malignant histiocytoses | ||
| H Group | Primary HLH: Monogenic inherited conditions leading to HLH | |
| Secondary HLH (non-Mendelian HLH) | ||
| HLH of unknown/uncertain origin | ||
LCH cells, known for many years to be a clonal proliferation, have now been shown to likely derive from a myeloid precursor whose proliferation is uniformly associated with activation of the MAPK/ERK signaling pathway.[5,6]
Clinically, LCH is a heterogenous disease that may involve a single organ (single-system LCH), which may be a single site (unifocal) or involve multiple sites (multifocal). It may also involve multiple organs (multisystem LCH). Multisystem LCH may involve a limited number of organs or be disseminated. Involvement of specific organs such as the liver, spleen, and hematopoietic system separates multisystem LCH into high-risk (multisystem risk-organ positive) and low-risk (multisystem risk-organ negative) groups, where risk indicates the risk of death from the disease.
References:
- Rodriguez-Galindo C, Allen CE: Langerhans cell histiocytosis. Blood 135 (16): 1319-1331, 2020.
- Emile JF, Abla O, Fraitag S, et al.: Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 127 (22): 2672-81, 2016.
- Berres ML, Lim KP, Peters T, et al.: BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med 211 (4): 669-83, 2014.
- Allen CE, Merad M, McClain KL: Langerhans-Cell Histiocytosis. N Engl J Med 379 (9): 856-868, 2018.
- Willman CL, Busque L, Griffith BB, et al.: Langerhans'-cell histiocytosis (histiocytosis X)--a clonal proliferative disease. N Engl J Med 331 (3): 154-60, 1994.
- Yu RC, Chu C, Buluwela L, et al.: Clonal proliferation of Langerhans cells in Langerhans cell histiocytosis. Lancet 343 (8900): 767-8, 1994.
Histopathological, Immunologic, and Cytogenetic Characteristics of LCH
Cell of Origin and Biological Correlates
The pathological histiocyte or Langerhans cell histiocytosis (LCH) cell has a gene expression profile closely resembling that of a myeloid dendritic cell. Studies have also demonstrated that the BRAF V600E variant can be identified in mononuclear cells in peripheral blood and cell-free DNA, usually in patients with disseminated disease.[1,2,3] This suggests that multisystem LCH arises from a somatic variant within the marrow or a circulating precursor cell, while localized disease arises from a variant occurring in a precursor cell at the local site.[2]
Modern classification of the histiocytic diseases subdivides them into dendritic cell–related, monocyte/macrophage-related, or true malignancies. LCH is a dendritic cell disease.[4,5] Comprehensive data analysis on gene expression array of LCH cells is consistent with the concept that the skin Langerhans cell (LC) is not the cell of origin for LCH.[1] Rather, the origin is likely to be a hematopoietic progenitor cell before being a committed myeloid dendritic cell, which expresses the same antigens (CD1a and CD207) as the skin LC.[6,7] This concept was further supported by reports that the transcription profile of LCH cells was distinct from myeloid and plasmacytoid dendritic cells, as well as epidermal LCs.[1,6,8,9]
LCH is now considered a myeloid neoplasm. However, some controversy remains as to whether it is a true malignancy or a neoplasm with varying clinical behavior. The same BRAF V600E variant has been found in many cancers; however, V600E-altered BRAF is also present in benign nevi, possibly indicating that malignant transformation requires additional variants.[10] These findings have raised the possibility of treatment with targeted therapies. Several trials of BRAF and MEK inhibitors are open for adults and children with LCH.
For more information, see the sections on Cytogenetic and Genomic Studies and Cytokine Analysis.
Histopathology
The Langerhans histiocytosis cells in LCH lesions (LCH cells) are immature dendritic cells, making up fewer than 10% of the cells present in the lesion.[9,11] These cells are classically large oval cells with abundant pink cytoplasm and a bean-shaped nucleus on hematoxylin and eosin stain. LCH cells stain positively with antibodies to S100, CD1a, and/or anti-Langerin (CD207). Staining with CD1a or Langerin confirms the diagnosis of LCH, but care should be taken to correlate with clinical presentation in organs in which normal LC cells occur.[12]
Because LCH cells activate other immunologic cells, LCH lesions also contain other histiocytes, lymphocytes, macrophages, neutrophils, eosinophils, and fibroblasts, and they may contain multinucleated giant cells.
In the brain, the following three types of histopathological findings have been described in LCH:
- Mass lesions in the meninges or choroid plexus with CD1a-positive LCH cells and predominantly CD8-positive lymphocytes.
- Mass lesions in connective tissue spaces with CD1a-positive LCH cells and predominantly CD8-positive lymphocytes that cause an inflammatory response and neuronal loss.
- Neurodegenerative lesions, consisting of cells staining for the altered BRAF protein with positive CD14, CD33, and CD163, identifying these as hematopoietic myeloid/monocytic cells. These are the pathological LCs that have migrated into the brain and do not stain with CD1a or CD207 and have become microglia-like.[13]
Immunologic Abnormalities
Normally, the LC is a primary presenter of antigen to naïve T lymphocytes. However, in LCH, the pathological dendritic cell does not efficiently stimulate primary T-lymphocyte responses.[14] Antibody staining for the dendritic cell markers, including CD80, CD86, and class II antigens, has shown that in LCH, the abnormal cells are immature dendritic cells. These cells present antigen poorly and are proliferating at a low rate.[11,14,15]
An expansion of regulatory T cells in patients with LCH has been reported.[15] The population of CD4-positive, CD25(high), FoxP3(high) cells was reported to comprise 20% of T cells and appeared to be in contact with LCH cells in the lesions. These T cells were present in peripheral blood in higher numbers in patients with LCH than in controls and returned to a normal level when patients were in remission.[15] Poorly functioning T cells expressing inhibitor receptors PD-1, TIM3, and LAG-3 have been found in LCH lesions but not in the peripheral blood of patients.[16] The dysfunctional T cells accumulate in LCH lesions, because PD-1 on the cell surface engages with the PD-L1 on the pathological dendritic cells.
Cytogenetic and Genomic Studies
Genomics of LCH
BRAF,NRAS, andARAFvariants
The genomic basis of LCH was advanced by a 2010 report of an activating variant of the BRAF oncogene (V600E) that was detected in 35 of 61 cases (57%).[17] Multiple subsequent reports have confirmed the presence of BRAF V600E variants in 50% or more of LCH cases in children.[2,18,19] Other BRAF variants that result in signal activation have been described.[18,20]ARAF variants are infrequent in LCH but, when present, can also lead to RAS-MAPK pathway activation.[21]
The presence of the BRAF V600E variant in blood and bone marrow was studied in a series of 100 patients, 65% of whom tested positive for the BRAF V600E variant by a sensitive quantitative polymerase chain reaction technique.[2] Circulating cells with the BRAF V600E variant could be detected in all high-risk patients and in a subset of low-risk multisystem patients. The BRAF V600E allele was detected in circulating cell-free DNA in 100% of patients with risk-organ–positive multisystem LCH, 42% of patients with risk-organ–negative LCH, and 14% of patients with single-system LCH.[22]
The myeloid dendritic cell origin of LCH was confirmed by finding CD34-positive stem cells with the variant in the bone marrow of high-risk patients. In those with low-risk disease, the variant was found in more mature myeloid dendritic cells, suggesting that the stage of cell development at which the somatic variant occurs is critical in defining the extent of disease in LCH.
Pulmonary LCH in adults was initially reported to be nonclonal in approximately 75% of cases,[23] while a later study of BRAF variants showed that 25% to 50% of adult patients with lung LCH had evidence of BRAF V600E variants.[23,24] Another study of 26 pulmonary LCH cases found that 50% had BRAF V600E variants and 40% had NRAS variants.[25] Approximately the same number of variants are polyclonal as are monoclonal. It has not been determined whether clonality and BRAF pathway variants are concordant in the same patients, which might suggest a reactive rather than a neoplastic condition in smoker's lung LCH and a clonal neoplasm in other types of LCH.
In a study of 117 patients with LCH, 83 adult patients with pulmonary LCH underwent molecular analysis. Nearly 90% of these patients had variants in the MAPK pathway.[26][Level of evidence C3] Of the 69 patients who had their biopsy samples further analyzed using a next-generation sequencing panel of 74 genes, 36% had BRAF V600E variants, 29% had BRAF N486-P490 deletions, 15% had MAP2K1 variants or deletions, and 4% had NRAS variants. Only one patient had a KRAS variant. Additionally, 11 patients had their biopsy samples analyzed using whole-exome sequencing. An average of 14 variants were found per patient, which is markedly higher than the average of one variant found per pediatric patient.[27] There were no clinical correlates, including presence of a BRAF V600E variant and smoking status. Of the 117 patients with LCH, 60% experienced a relapse.
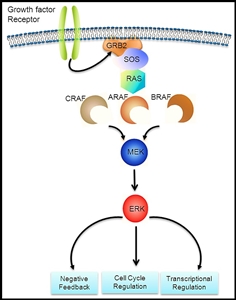
Figure 1. Courtesy of Rikhia Chakraborty, Ph.D. Permission to reuse the figure in any form must be obtained directly from Dr. Chakraborty.
The RAS-MAPK signaling pathway (see Figure 1) transmits signals from a cell surface receptor (e.g., a growth factor) through the RAS pathway (via one of the RAF proteins [A, B, or C]) to phosphorylate MEK and then the extracellular signal-regulated kinase (ERK), which leads to nuclear signals affecting cell cycle and transcription regulation. The V600E variant of BRAF leads to continuous phosphorylation, and thus activation, of MEK and ERK without the need for an external signal. Activation of ERK occurs by phosphorylation, and phosphorylated ERK can be detected in virtually all LCH lesions.[17,28]
In a mouse model of LCH, the BRAF V600E variant was shown to inhibit a chemokine receptor (CCR7)–mediated migration of dendritic cells, forcing them to accumulate in the LCH lesion.[29] This variant also causes an increased expression of BCL2L1, which results in resistance to apoptosis. This process leads to the cells being less responsive to chemotherapy. The BRAF V600E variant also causes growth arrest of hematopoietic progenitor cells and a senescence-associated secretory phenotype that further promotes accumulation of the pathological cells.[30]
Another mouse model with the BRAF V600E variant under control of Scl or Map17 gene promoters added additional insights into the biology of neurodegenerative LCH.[31] These studies confirmed the hematopoietic origin of CD11a-positive macrophages with BRAF V600E variants. This process disrupts the blood-brain barrier and causes loss of Purkinje cells and progressive neurodegeneration by resistance to apoptosis and production of senescent associated secretory proteins, which include inflammatory cytokines IL-1, IL-6, and matrix metalloproteinases. Treatment with a MAP kinase inhibitor and a senolytic agent (navitoclax) decreased the pathogenic cell numbers and led to clinical improvement in the mice.
In summary, LCH is now considered a myeloid neoplasm primarily driven by activating variants of the MAPK pathway. Fifty percent to 60% of the activating variants are caused by BRAF V600E variants, which are enriched in patients with multisystem risk organ–positive LCH and in patients with neurodegenerative-disease LCH.[32] Ongoing studies are assessing whether low-level variant detection in peripheral blood can be used as a minimal residual disease marker to assist in therapeutic decisions.
Other RAS-MAPK pathway alterations
Because RAS-MAPK pathway activation (elevated phosphor-ERK) can be detected in all LCH cases, including those without BRAF variants, the presence of genomic alterations in other components of the pathway was suspected. The following genomic alterations were identified:
-
MAP2K1 variants. Whole-exome sequencing on biopsy samples of BRAF-altered versus BRAF–wild-type LCH tissue revealed that 7 of 21 BRAF–wild-type specimens had MAP2K1 variants, while no BRAF-altered specimens had MAP2K1 variants.[28] The variants in MAP2K1 (which codes for MEK1) were activating, as indicated by their induction of ERK phosphorylation.[28]
Another study showed MAP2K1 variants exclusively in 11 of 22 BRAF–wild-type cases.[33] One study showed that MAP2K1 and other variants associated with pediatric and adult LCH were mutually exclusive of BRAF variants.[34] The authors found a variety of variants in other pathways (e.g., JNK, RAS-ERK, and JAK-STAT) in pediatric and adult patients with BRAF V600E or MAP2K1 variants. Another study evaluated the kinase alterations and myeloid-associated variants in 73 adult patients with LCH.[35] They reported a median of two variants per adult patient, as opposed to children who usually have only one variant. BRAF V600E was found in 31%, BRAF indel in 29%, and MAP2K1 in 19% of patients with LCH. A variety of other protein kinase and related pathways were found in 89% of adult patients with LCH. MAP2K1 variants were exclusive of BRAF variants.
- In-frame BRAF deletions and FAM73A::BRAF gene fusions. In-frame BRAF deletions and in-frame FAM73A::BRAF gene fusions have occurred in the group of BRAF V600E and MAP2K1 variant–negative cases.[27]
In summary, studies support the universal activation of ERK in LCH. ERK activation in most cases of LCH is explained by BRAF and MAP2K1 alterations.[17,27,28] Altogether, these variants in the MAP kinase pathway account for nearly 80% of the causes of the universal activation of ERK in LCH.[17,27,28] The remaining cases have a range of variants that include small deletions in BRAF, BRAF gene fusions (discussed above), as well as variants in ARAF, MAP3K1, NRAS, ERBB3, PI3CA, CSF1R, and other rare targets.[34,32][Level of evidence C1]
Clinical implications
Clinical implications of the described genomic findings include the following:
- LCH is included in a group of other pediatric tumors with activating BRAF variants, such as select nonmalignant conditions (e.g., benign nevi) [36] and low-grade malignancies (e.g., pilocytic astrocytoma).[37,38] All of these conditions have a generally indolent course, with spontaneous resolution occurring in some cases. This distinctive clinical course may be a manifestation of oncogene-induced senescence.[36,39]
- In some pediatric studies, BRAF V600E variants have been associated with more severe multisystem disease, treatment failure, increased reactivations, and an increased risk of neurodegeneration (see below).[40] These clinical correlates were recently investigated for non-BRAF V600E variants in an international study. Similar to the BRAF V600E cohort, all patients with multisystem risk organ–positive LCH had detectable variants in peripheral blood mononuclear cells. Of seven patients with multisystem risk organ–negative LCH, four had detectable variants. No patients with single-system disease had detectable variants in peripheral blood mononuclear cells. The authors concluded that other MAPK pathway variants are associated with risk status, similar to BRAF V600E variants.[32]
BRAF V600E variants can be targeted by BRAF inhibitors (e.g., vemurafenib and dabrafenib) or by the combination of BRAF inhibitors plus MEK inhibitors (e.g., dabrafenib/trametinib and vemurafenib/cobimetinib). These agents and combinations are approved for adults with melanoma. Treatment of melanoma in adults with combinations of a BRAF inhibitor and a MEK inhibitor showed significantly improved progression-free survival outcomes compared with treatment using a BRAF inhibitor alone.[41,42]
Several case reports and two case series have also demonstrated the efficacy of BRAF inhibitors for the treatment of LCH in children.[43,44,45,46,47,48] However, the long-term role of this therapy is complicated because most patients will relapse when the inhibitors are discontinued. For more information, see the sections on Treatment of recurrent, refractory, or progressive high-risk disease: multisystem LCH and Targeted therapies for the treatment of single-system and multisystem disease.
- Circulating BRAF V600E–altered cells have been found in 59% of patients who developed neurodegenerative-disease LCH, compared with 15% of patients who did not develop neurodegenerative-disease LCH. Detectable altered circulating cells had a sensitivity of 0.59 and specificity of 0.86 for developing the neurodegenerative disease. Even after therapy, some patients with neurodegenerative-disease LCH had circulating BRAF V600E–altered cells.[13]
- With additional research, the observation of the BRAF V600E variant (or potentially MAP2K1 variants) in circulating cells or cell-free DNA may become a useful diagnostic tool to define high-risk versus low-risk disease.[2] Additionally, for patients who have a somatic variant, persistence of circulating cells with the variant may be useful as a marker of residual disease.[2]
Cytokine Analysis
Immunohistochemical staining has shown upregulation of many different cytokines/chemokines, both in LCH lesions and in the serum/plasma of patients with LCH.[49,50] In an analysis of gene expression in LCH by gene array techniques, 2,000 differentially expressed genes were identified. Of 65 genes previously reported to be associated with LCH, only 11 were found to be upregulated in the array results. The most highly upregulated gene in both CD207-positive and CD3-positive cells was SPP1 (encoding the osteopontin protein); other genes that activate and recruit T cells to sites of inflammation are also upregulated.[1] The expression profile of the T cells was that of an activated regulatory T-cell phenotype with increased expression of FOXP3, CTLA4, and SPP1. These findings support a previous report on the expansion of regulatory T cells in LCH.[1] There was pronounced expression of genes associated with early myeloid progenitors such as CD33 and CD44, which is consistent with an earlier report of elevated myeloid dendritic cells in the blood of patients with LCH.[51] A model of Misguided Myeloid Dendritic Cell Precursors has been proposed, whereby myeloid dendritic cell precursors are recruited to sites of LCH by an unknown mechanism, and the dendritic cells, in turn, recruit lymphocytes by excretion of osteopontin, neuropilin-1, and vannin-1.[1]
One study evaluated possible biomarkers for central nervous system LCH. The study examined 121 unique proteins in the cerebrospinal fluid (CSF) of 40 pediatric patients with LCH and compared them with controls, which included 29 patients with acute lymphoblastic leukemia, 25 patients with brain tumors, 28 patients with neurodegenerative diseases, and 9 patients with hemophagocytic lymphohistiocytosis. Only osteopontin proved to be significantly increased in the CSF of LCH patients with either neurodegeneration or mass lesions (pituitary), compared with all of the control groups. Analysis of osteopontin expression in these tissues confirmed an upregulation of the SPP1 gene.[13]
Several investigators have published studies evaluating the level of various cytokines or growth factors in the blood of patients with LCH. These studies have included many of the genes found not to be upregulated by the gene expression results discussed above.[1] One explanation for elevated levels of these proteins is a systemic inflammatory response, with the cytokines/growth factors being produced by cells outside the LCH lesions. A second possible explanation is that macrophages in the LCH lesions produce the cytokines measured in the blood or are concentrated in lesions.
IL-1 beta and prostaglandin GE2 levels were measured in the saliva of patients with oral LCH lesions or multisystem high-risk patients with and without oral lesions. Levels of both were higher in patients with active disease and decreased after successful therapy.[52]
References:
- Allen CE, Li L, Peters TL, et al.: Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profile compared with epidermal Langerhans cells. J Immunol 184 (8): 4557-67, 2010.
- Berres ML, Lim KP, Peters T, et al.: BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med 211 (4): 669-83, 2014.
- Hyman DM, Diamond EL, Vibat CR, et al.: Prospective blinded study of BRAFV600E mutation detection in cell-free DNA of patients with systemic histiocytic disorders. Cancer Discov 5 (1): 64-71, 2015.
- Emile JF, Abla O, Fraitag S, et al.: Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 127 (22): 2672-81, 2016.
- Picarsic J, Jaffe R: Nosology and Pathology of Langerhans Cell Histiocytosis. Hematol Oncol Clin North Am 29 (5): 799-823, 2015.
- Ginhoux F, Merad M: Ontogeny and homeostasis of Langerhans cells. Immunol Cell Biol 88 (4): 387-92, 2010 May-Jun.
- Durham BH, Roos-Weil D, Baillou C, et al.: Functional evidence for derivation of systemic histiocytic neoplasms from hematopoietic stem/progenitor cells. Blood 130 (2): 176-180, 2017.
- Hutter C, Kauer M, Simonitsch-Klupp I, et al.: Notch is active in Langerhans cell histiocytosis and confers pathognomonic features on dendritic cells. Blood 120 (26): 5199-208, 2012.
- Berres ML, Allen CE, Merad M: Pathological consequence of misguided dendritic cell differentiation in histiocytic diseases. Adv Immunol 120: 127-61, 2013.
- Badalian-Very G, Vergilio JA, Fleming M, et al.: Pathogenesis of Langerhans cell histiocytosis. Annu Rev Pathol 8: 1-20, 2013.
- Geissmann F, Lepelletier Y, Fraitag S, et al.: Differentiation of Langerhans cells in Langerhans cell histiocytosis. Blood 97 (5): 1241-8, 2001.
- Chikwava K, Jaffe R: Langerin (CD207) staining in normal pediatric tissues, reactive lymph nodes, and childhood histiocytic disorders. Pediatr Dev Pathol 7 (6): 607-14, 2004 Nov-Dec.
- McClain KL, Picarsic J, Chakraborty R, et al.: CNS Langerhans cell histiocytosis: Common hematopoietic origin for LCH-associated neurodegeneration and mass lesions. Cancer 124 (12): 2607-2620, 2018.
- Yu RC, Morris JF, Pritchard J, et al.: Defective alloantigen-presenting capacity of 'Langerhans cell histiocytosis cells'. Arch Dis Child 67 (11): 1370-2, 1992.
- Senechal B, Elain G, Jeziorski E, et al.: Expansion of regulatory T cells in patients with Langerhans cell histiocytosis. PLoS Med 4 (8): e253, 2007.
- Sengal A, Velazquez J, Hahne M, et al.: Overcoming T-cell exhaustion in LCH: PD-1 blockade and targeted MAPK inhibition are synergistic in a mouse model of LCH. Blood 137 (13): 1777-1791, 2021.
- Badalian-Very G, Vergilio JA, Degar BA, et al.: Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 116 (11): 1919-23, 2010.
- Satoh T, Smith A, Sarde A, et al.: B-RAF mutant alleles associated with Langerhans cell histiocytosis, a granulomatous pediatric disease. PLoS One 7 (4): e33891, 2012.
- Sahm F, Capper D, Preusser M, et al.: BRAFV600E mutant protein is expressed in cells of variable maturation in Langerhans cell histiocytosis. Blood 120 (12): e28-34, 2012.
- Héritier S, Hélias-Rodzewicz Z, Chakraborty R, et al.: New somatic BRAF splicing mutation in Langerhans cell histiocytosis. Mol Cancer 16 (1): 115, 2017.
- Nelson DS, Quispel W, Badalian-Very G, et al.: Somatic activating ARAF mutations in Langerhans cell histiocytosis. Blood 123 (20): 3152-5, 2014.
- Héritier S, Hélias-Rodzewicz Z, Lapillonne H, et al.: Circulating cell-free BRAF(V600E) as a biomarker in children with Langerhans cell histiocytosis. Br J Haematol 178 (3): 457-467, 2017.
- Dacic S, Trusky C, Bakker A, et al.: Genotypic analysis of pulmonary Langerhans cell histiocytosis. Hum Pathol 34 (12): 1345-9, 2003.
- Roden AC, Hu X, Kip S, et al.: BRAF V600E expression in Langerhans cell histiocytosis: clinical and immunohistochemical study on 25 pulmonary and 54 extrapulmonary cases. Am J Surg Pathol 38 (4): 548-51, 2014.
- Mourah S, How-Kit A, Meignin V, et al.: Recurrent NRAS mutations in pulmonary Langerhans cell histiocytosis. Eur Respir J 47 (6): 1785-96, 2016.
- Jouenne F, Chevret S, Bugnet E, et al.: Genetic landscape of adult Langerhans cell histiocytosis with lung involvement. Eur Respir J 55 (2): , 2020.
- Chakraborty R, Burke TM, Hampton OA, et al.: Alternative genetic mechanisms of BRAF activation in Langerhans cell histiocytosis. Blood 128 (21): 2533-2537, 2016.
- Chakraborty R, Hampton OA, Shen X, et al.: Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood 124 (19): 3007-15, 2014.
- Hogstad B, Berres ML, Chakraborty R, et al.: RAF/MEK/extracellular signal-related kinase pathway suppresses dendritic cell migration and traps dendritic cells in Langerhans cell histiocytosis lesions. J Exp Med 215 (1): 319-336, 2018.
- Bigenwald C, Le Berichel J, Wilk CM, et al.: BRAFV600E-induced senescence drives Langerhans cell histiocytosis pathophysiology. Nat Med 27 (5): 851-861, 2021.
- Wilk CM, Cathomas F, Török O, et al.: Circulating senescent myeloid cells infiltrate the brain and cause neurodegeneration in histiocytic disorders. Immunity 56 (12): 2790-2802.e6, 2023.
- Milne P, Abhyankar H, Scull B, et al.: Cellular distribution of mutations and association with disease risk in Langerhans cell histiocytosis without BRAFV600E. Blood Adv 6 (16): 4901-4904, 2022.
- Brown NA, Furtado LV, Betz BL, et al.: High prevalence of somatic MAP2K1 mutations in BRAF V600E-negative Langerhans cell histiocytosis. Blood 124 (10): 1655-8, 2014.
- Durham BH, Lopez Rodrigo E, Picarsic J, et al.: Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med 25 (12): 1839-1842, 2019.
- Chen J, Zhao AL, Duan MH, et al.: Diverse kinase alterations and myeloid-associated mutations in adult histiocytosis. Leukemia 36 (2): 573-576, 2022.
- Michaloglou C, Vredeveld LC, Soengas MS, et al.: BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436 (7051): 720-4, 2005.
- Jones DT, Kocialkowski S, Liu L, et al.: Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68 (21): 8673-7, 2008.
- Pfister S, Janzarik WG, Remke M, et al.: BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest 118 (5): 1739-49, 2008.
- Jacob K, Quang-Khuong DA, Jones DT, et al.: Genetic aberrations leading to MAPK pathway activation mediate oncogene-induced senescence in sporadic pilocytic astrocytomas. Clin Cancer Res 17 (14): 4650-60, 2011.
- Héritier S, Emile JF, Barkaoui MA, et al.: BRAF Mutation Correlates With High-Risk Langerhans Cell Histiocytosis and Increased Resistance to First-Line Therapy. J Clin Oncol 34 (25): 3023-30, 2016.
- Larkin J, Ascierto PA, Dréno B, et al.: Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 371 (20): 1867-76, 2014.
- Long GV, Stroyakovskiy D, Gogas H, et al.: Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet 386 (9992): 444-51, 2015.
- Eckstein OS, Visser J, Rodriguez-Galindo C, et al.: Clinical responses and persistent BRAF V600E+ blood cells in children with LCH treated with MAPK pathway inhibition. Blood 133 (15): 1691-1694, 2019.
- Donadieu J, Larabi IA, Tardieu M, et al.: Vemurafenib for Refractory Multisystem Langerhans Cell Histiocytosis in Children: An International Observational Study. J Clin Oncol 37 (31): 2857-2865, 2019.
- Kolenová A, Schwentner R, Jug G, et al.: Targeted inhibition of the MAPK pathway: emerging salvage option for progressive life-threatening multisystem LCH. Blood Adv 1 (6): 352-356, 2017.
- Lee LH, Gasilina A, Roychoudhury J, et al.: Real-time genomic profiling of histiocytoses identifies early-kinase domain BRAF alterations while improving treatment outcomes. JCI Insight 2 (3): e89473, 2017.
- Héritier S, Jehanne M, Leverger G, et al.: Vemurafenib Use in an Infant for High-Risk Langerhans Cell Histiocytosis. JAMA Oncol 1 (6): 836-8, 2015.
- Váradi Z, Bánusz R, Csomor J, et al.: Effective BRAF inhibitor vemurafenib therapy in a 2-year-old patient with sequentially diagnosed Langerhans cell histiocytosis and Erdheim-Chester disease. Onco Targets Ther 10: 521-526, 2017.
- Fleming MD, Pinkus JL, Fournier MV, et al.: Coincident expression of the chemokine receptors CCR6 and CCR7 by pathologic Langerhans cells in Langerhans cell histiocytosis. Blood 101 (7): 2473-5, 2003.
- Annels NE, Da Costa CE, Prins FA, et al.: Aberrant chemokine receptor expression and chemokine production by Langerhans cells underlies the pathogenesis of Langerhans cell histiocytosis. J Exp Med 197 (10): 1385-90, 2003.
- Rolland A, Guyon L, Gill M, et al.: Increased blood myeloid dendritic cells and dendritic cell-poietins in Langerhans cell histiocytosis. J Immunol 174 (5): 3067-71, 2005.
- Preliasco VF, Benchuya C, Pavan V, et al.: IL-1 beta and PGE2 levels are increased in the saliva of children with Langerhans cell histiocytosis. J Oral Pathol Med 37 (9): 522-7, 2008.
Childhood LCH
General Information About Childhood LCH
Incidence
The annual incidence of Langerhans cell histiocytosis (LCH) has been estimated to be between two and ten cases per 1 million children aged 15 years or younger.[1,2,3] The male-to-female ratio (M:F) is close to one, and the median age of presentation is 30 months.[4] A 4-year survey of 251 new LCH cases in France found an annual incidence of 4.6 cases per 1 million children younger than 15 years (M:F, 1.2).[5]
A population-based study identified 658 patients with LCH who were diagnosed in England from 2013 to 2019.[6] The prevalence of LCH was 9.95 cases per 1 million people at the end of 2019. Forty-nine percent of patients were younger than 15 years, with an incidence rate of 4.46 cases per 1 million children per year. The authors felt that this incidence is likely an underestimate, particularly for single-system LCH. This is the first study to accurately identify adult patients aged 30 years to 60 years and older. However, the study also included patients aged 15 to 29 years in the adult category, which resulted in a total adult incidence rate of 1.06 cases per 1 million adults per year. Patients living in lower socioeconomic circumstances and those older than 30 years had worse survival rates than those of higher socioeconomic status or children.
Surveillance, Epidemiology, and End Results (SEER) registry data from 2000 to 2009 were reviewed to identify high-risk LCH cases and assess demographic variables.[7] Of 145 cases, the age-standardized incidence for disseminated disease was 0.7 per 1 million children per year, with lower incidence in Black patients (0.41 per 1 million) and higher incidence in Hispanic patients (1.63 per 1 million) younger than 5 years. Crowded living conditions and lower socioeconomic circumstances were associated with a higher risk of LCH, possibly because of the correlation with maternal and neonatal infections.[8] In a population-based, case-control study, Hispanic mothers were more likely than non-Hispanic White mothers to have children who developed LCH; this risk increased when both parents were Hispanic. Non-Hispanic Black mothers were less likely than non-Hispanic White mothers to give birth to children who developed LCH.[9] In addition, a family-based genome-wide association study found that a polymorphism of the SMAD6 gene was highly associated with LCH, especially in Hispanic patients.[10] The study from England (described above) included 658 adults and children, 79% of whom were White. This study did not show an increased incidence in the Hispanic population, reflecting the differences in the U.K. population.[6]
Risk factors
Although the following risk factors have been proposed for LCH, strong and consistent associations have not been confirmed:
- Parental exposure to solvents.[8]
- Family history of cancer.[11]
- Personal or family history of thyroid disease.[8,12]
- Perinatal infections.[8,11]
- Parental occupational exposure to metal, granite, or wood dust.[11]
- Hispanic ethnicity and race.[7]
- Low socioeconomic status.[7]
- Lack of childhood vaccinations.[11]
Efforts to define a viral cause have not been successful.[13,14]
Diagnostic evaluation
The complete evaluation of any patient presenting with LCH includes the following:[15]
- History and physical examination: A complete history and physical examination with special attention to the skin, lymph nodes, ears, oral pharynx, gingiva, tongue, teeth, bones, lungs, thyroid, liver and spleen size, bone abnormalities, growth velocity, and history of excessive thirst and urination.
Other tests and procedures include the following:
-
Blood tests: Blood tests include complete blood count with leukocyte differential and platelet count, liver function tests (e.g., bilirubin, albumin, aspartate aminotransferase, alanine aminotransferase, gamma glutamyl transferase, and prothrombin time or international normalized ratio (INR)/partial thromboplastin time in patients with hepatomegaly, jaundice, elevations of liver enzymes, or low albumin), and serum electrolytes.
In patients with severe multisystem LCH, additional tests for secondary hemophagocytic lymphohistiocytosis such as ferritin, triglycerides, fibrinogen, d-dimers, lactate dehydrogenase, CXCL9, and sCD25, may be indicated.
- Assessment of the RAS-RAF-MEK pathway: Although assessment of the RAS-RAF-MEK pathway is not a required part of the workup for patients with LCH, the BRAF variant can be detected by either immunohistochemistry or molecular diagnostic methods in fresh tissue, formalin-fixed tissue, and peripheral blood.
- Urine tests: Urine tests include urinalysis and a water-deprivation test if diabetes insipidus is suspected. Water deprivation tests in very young children, especially infants, are performed under close medical monitoring.
- Bone marrow aspirate and biopsy: A bone marrow aspirate and biopsy is indicated for patients with multisystem disease who have unexplained anemia or thrombocytopenia. The biopsy specimens should be stained with anti-CD1a and/or anti-CD207 (langerin) and anti-CD163 immunostains to facilitate the detection of LCH cells. Polymerase chain reaction (PCR) analysis for BRAF-altered cells is also important.
-
Radiological and imaging tests: Radiological tests for the first level of screening include skeletal survey, skull series, bone scans, and chest X-ray. Positron emission tomography (PET) scans are becoming more widely used because of superior diagnostic index and evaluation of response to therapy compared with bone scans.[16,17,18]
- Computed tomography (CT) scan: CT scan of the head may be indicated if orbital, mastoid, or other maxillofacial involvement is suspected. Imaging tests may include magnetic resonance imaging (MRI) scan with gadolinium contrast of the brain for patients with diabetes insipidus or suspected brain or vertebral involvement.[19]
CT scan of the lungs may be indicated for patients with abnormal chest X-rays or pulmonary symptoms. High-resolution CT scans may show evidence of pulmonary LCH when the chest X-ray is normal. Thus, in infants and toddlers with normal chest X-rays, a CT scan may be considered when respiratory signs or symptoms are present. Patients with pulmonary LCH may also have normal chest X-rays and abnormal pulmonary function tests.[20]
LCH causes fatty changes in the liver or hypodense areas along the portal tract, which can be identified by CT scan, if indicated.[21]
- Fluorine F 18-fludeoxyglucose (18F-FDG) PET scan: 18F-FDG PET scan abnormalities were reported in the brains of seven patients with LCH who exhibited neurological and radiographic signs of neurodegenerative disease.[18] There was good correlation with MRI findings in the cerebellar white matter, but less so in the caudate nuclei and frontal cortex. It was suggested that PET scans of patients at high risk of developing neurodegenerative LCH could show abnormalities earlier than MRI.[18] PET scans often demonstrate lesions not found by other modalities and show a decrease of activity of LCH after 6 weeks of therapy, providing a better assessment of response to therapy than bone scans or plain x-rays.[17,22] However, one study suggests that bone scans are more sensitive than PET scans for lesions in the hands and feet.[23]
- PET-CT scan.[24]
- MRI: MRI findings in patients with diabetes insipidus include thickening and nodularity of the pituitary stalk with loss of the posterior pituitary bright spot, reflecting absence of antidiuretic hormone.
All patients with vertebral body involvement need careful assessment of associated soft tissue, which may impinge on the spinal cord.
MRI findings of central nervous system (CNS) LCH include T2 FLAIR enhancement in the pons, basal ganglia, white matter of the cerebellum, and mass lesions or meningeal enhancement. In a report of 163 patients, meningeal lesions were found in 29% of patients and choroid plexus involvement was found in 6% of patients. Paranasal sinus or mastoid lesions were found in 55% of patients versus 20% of controls, and accentuated Virchow-Robin spaces were found in 70% of patients versus 27% of controls.[25]
- Computed tomography (CT) scan: CT scan of the head may be indicated if orbital, mastoid, or other maxillofacial involvement is suspected. Imaging tests may include magnetic resonance imaging (MRI) scan with gadolinium contrast of the brain for patients with diabetes insipidus or suspected brain or vertebral involvement.[19]
-
Biopsy: Lytic bone lesions, skin, and lymph nodes are the sites most frequently biopsied for diagnosis of LCH. A liver biopsy is indicated when a child with LCH presents with hypoalbuminemia not caused by gastrointestinal LCH or another etiology. These patients usually have elevated levels of bilirubin or liver enzymes. An open lung biopsy may be necessary for obtaining tissue for diagnosis of pulmonary LCH when bronchoalveolar lavage is nondiagnostic. Diagnosing gastrointestinal involvement with LCH is difficult because of patchy involvement. Careful endoscopic examination that includes multiple biopsies is usually needed.
A pathological diagnosis is always required to make a definitive diagnosis. However, this may sometimes be difficult or contraindicated, such as in isolated pituitary stalk disease or vertebra plana without a soft tissue mass, when the risk outweighs the benefit of a firm diagnosis.
Prognostic factors
Survival is closely linked to the extent of disease at presentation when high-risk organs (liver, spleen, and/or bone marrow) are involved, as well as the response to initial treatment. Many studies have confirmed the high mortality rate (35%) in patients with high-risk multisystem disease, when they do not respond well to therapy in the first 6 weeks.[26] Because of treatment advances, including early implementation of additional therapy for poor responders, the outcome for children with LCH involving high-risk organs has improved.[27,28] Data from HISTSOC-LCH-III (NCT00276757) showed an overall survival (OS) rate of 84% for patients treated for 12 months with systemic chemotherapy.[29]
For many years, lungs were thought to be high-risk organs, but isolated lung involvement in pediatric LCH is no longer considered to pose a significant risk of death,[26] unless pneumothorax or bilateral pneumothoraces occur.
Patients with single-system disease and low-risk multisystem disease do not usually die of LCH, but recurrent disease may result in considerable morbidity and significant late effects.[30] Overall, recurrences have been found in 10% of patients with single-system unifocal disease, 25% of patients with single-system multifocal bone LCH, and 50% of patients with low-risk multisystem disease and those with high-risk multisystem disease who achieve nonactive disease status with chemotherapy. HISTSOC-LCH-III data showed a significant difference in reactivation rate for low–risk-organ patients randomly assigned to receive 6 months of treatment (54%) versus 12 months of treatment (37%).[29] Similarly, the nonrandomized high-risk group of patients who were all treated for 12 months had a reactivation rate of 30%, compared with more than 50% in previous studies in which patients were treated with the same therapy for 6 months.[29]
Most high-risk patients whose disease reactivated (30%) after achieving a no active disease (NAD) status will do so in low-risk organs such as bone. These patients will have the same risk of late effects as patients with low-risk multisystem disease.[29] The major current treatment challenge is to reduce this overall 20% to 30% incidence of reactivations and the significant risk of serious permanent consequences in this group of patients.
Apart from disease extent, prognostic factors for children with LCH include the following:
- Age at diagnosis. Although age younger than 2 years was once thought to portend a worse prognosis, data from the HISTSOC-LCH-II study showed that patients aged 2 years or younger without high–risk-organ involvement had the same response to therapy as did older patients.[28] In contrast, the OS was poorer in neonates with risk-organ involvement compared with infants and children with the same extent of disease when patients were treated for only 6 months.[28]
- Response to treatment. Response to therapy at 6 to 12 weeks has been shown to be a more important prognostic factor than age.[31] The overall response to therapy is influenced by the duration and intensity of treatment.[27,28]
- Site of involvement.
-
BRAF or MAP2K1 variants.
A study of 173 patients with the BRAF V600E variant and 142 without the variant revealed that the variant occurred in 88% of patients with high-risk disease, 69% of patients with multisystem low-risk LCH, and 44% of patients with single-system low-risk LCH.[32] The variant was also found in 75% of patients with the neurodegenerative syndrome and 73% of patients with pituitary involvement. The BRAF V600E variant was also associated with an increased incidence of skin disease and a younger age of presentation. Resistance to initial treatment and relapse were higher in patients with the variant. MAP2K1 variants were associated with single-system bone disease.[32]
An earlier study of 100 patients did not find all these clinical correlations, except that relapses occurred more frequently in patients with low-risk and high-risk LCH and the BRAF V600E variant.[33]
An international collaborative study of 377 patients found 300 patients (79.6%) with MAPK pathway variants and compared them with patients without variants. This study confirmed the findings of a previous study. It also found an increased risk of CNS-risk bone LCH, gastrointestinal and skin involvement, and fewer cases of BRAF-positive single-system, multifocal bone LCH among patients with MAPK pathway variants.[34] A cohort of patients with the BRAF exon 12 variant had a higher incidence of lung LCH. MAP2K1 variants were more frequent in patients with single-system bone LCH, but not in patients with CNS-risk bone LCH. The prognostic impact of the BRAF variant was more strongly associated with having risk-organ and multisystem involvement, rather than the presence of the variant itself.
A significant proportion of patients who survive LCH experience disease relapses and/or develop permanent conditions. Central diabetes insipidus is the most common condition, and CNS neurodegenerative LCH is the most severe condition.[35]
Follow-up considerations in childhood LCH
Because of the risk of reactivation (which ranges from 10% in single-system unifocal bone lesions to close to 50% in low-risk and high-risk multisystem LCH) and the risk of permanent long-term effects, LCH patients need to be monitored for many years.
Patients with diabetes insipidus and/or skull lesions in the orbit, mastoid, or temporal bones appear to be at higher risk of LCH CNS involvement and LCH CNS neurodegenerative syndrome. These patients should have MRI scans with gadolinium contrast at the time of LCH diagnosis and every 1 to 2 years thereafter for 10 years to detect evidence of CNS disease.[36] The Histiocyte Society CNS LCH Committee does not recommend any treatment for radiological CNS LCH of the neurodegenerative type if there is no associated clinical neurodegeneration and the MRI findings remain stable. However, careful neurological examinations and appropriate imaging with MRI are suggested at regular intervals.[37]
Auditory brain-stem response tests should be done at regular intervals to define the onset of clinical CNS LCH as early as possible, as this may affect response to therapy.[38] When clinical signs are present, intervention is indicated in patients with radiological evidence of LCH-associated changes in the cerebellum. Available studies of different forms of therapy for CNS neurodegeneration suggest that the neurodegenerative changes may be stabilized or improved, but only if therapy is started early.[38] It is critical to monitor patients at risk with neurological examinations and serial brain MRI scans. For more information, see the Clinical neurodegenerative syndrome LCH (cND-LCH) section.
For children with LCH in the lung, pulmonary function testing and chest CT scans are sensitive methods for detecting disease progression.[39]
A 16-year follow-up study of patients from one institution suggested that children with LCH have an increased risk of developing adult smoker's lung LCH compared with normal young adults who smoke. Ongoing re-education regarding this risk should be part of the routine follow-up of children with LCH at any site.[39]
In summary, many patients with multisystem disease will experience long-term sequelae caused by their underlying disease and/or treatment. Endocrine and CNS sequelae are the most common. These long-term sequelae significantly affect health-related quality of life in many of these patients.[40][Level of evidence C1] Specific long-term follow-up guidelines after treatment of childhood cancer or other conditions with chemotherapy have been published by the Children's Oncology Group and are available on their website. For more information, see the Late Disease and Treatment Effects of Childhood LCH section.
Special Considerations for the Treatment of Children With Cancer
Cancer in children and adolescents is rare, although the overall incidence has slowly increased since 1975.[41] Children and adolescents with cancer should be referred to medical centers that have a multidisciplinary team of cancer specialists with experience treating the cancers that occur during childhood and adolescence. This multidisciplinary team approach incorporates the skills of the following pediatric specialists and others to ensure that children receive treatment, supportive care, and rehabilitation to achieve optimal survival and quality of life:
- Primary care physicians.
- Pediatric surgeons.
- Transplant surgeons.
- Pathologists.
- Pediatric radiation oncologists.
- Pediatric medical oncologists and hematologists.
- Ophthalmologists.
- Rehabilitation specialists.
- Pediatric oncology nurses.
- Social workers.
- Child-life professionals.
- Psychologists.
- Nutritionists.
For specific information about supportive care for children and adolescents with cancer, see the summaries on Supportive and Palliative Care.
The American Academy of Pediatrics has outlined guidelines for pediatric cancer centers and their role in the treatment of children and adolescents with cancer.[42] At these centers, clinical trials are available for most types of cancer that occur in children and adolescents, and the opportunity to participate is offered to most patients and their families. Clinical trials for children and adolescents diagnosed with cancer are generally designed to compare potentially better therapy with current standard therapy. Other types of clinical trials test novel therapies when there is no standard therapy for a cancer diagnosis. Most of the progress in identifying curative therapies for childhood cancers has been achieved through clinical trials. Information about ongoing clinical trials is available from the NCI website.
Low-Risk Disease: Single-System or Multisystem LCH
Clinical presentation of low-risk, single-system or multisystem LCH
LCH most commonly presents as a painful bone lesion, with skin being the second most commonly involved organ. Systemic symptoms of fever, weight loss, diarrhea, edema, dyspnea, polydipsia, and polyuria relate to specific organ involvement and single-system or multisystem disease presentation (see Table 2).[35]
| Clinical Group | Description | ||
|---|---|---|---|
| CNS = central nervous system; LACI = LCH-associated abnormal CNS imaging; LACS = LCH-associated abnormal CNS symptoms; LCH = Langerhans cell histiocytosis. | |||
| a Reprinted fromBlood, Volume 135, Issue 16, Carlos Rodriguez-Galindo, Carl E. Allen, Langerhans cell histiocytosis, Pages 1319–1331, Copyright 2020, with permission from Elsevier.[35] | |||
| Multisystem | Two or more systems involved | ||
| With risk-organ involvement | Involvement of liver, spleen, or bone marrow | ||
| Without risk-organ involvement | Without involvement of liver, spleen, or bone marrow | ||
| Single-system | Only one system involved | ||
| Single site | Skin, bone, lymph node, other (thyroid, thymus) | ||
| Multiple sites | Multifocal bone disease | ||
| Special site | Skull-base lesion with intracranial extension or vertebral lesion with intraspinal soft tissue extension | ||
| Pulmonary LCH | Isolated lung disease | ||
| CNS LCH | Tumorous lesions | ||
| Neurodegenerative disease | |||
| LACI | |||
| LACS | |||
Specific organs are considered high risk or low risk when involved at disease presentation. Risk refers to the risk of mortality in high-risk patients. Chronic recurrent involvement of low-risk organs, while usually not life-threatening, can result in potentially devastating long-term consequences.
- High-risk organs include the liver, spleen, and hematopoietic system (defined by the presence of at least two lineage abnormalities in blood or by pathological CD1a-positive or CD207-positive cells in the bone marrow). Newer technologies (BRAF V600E detection PCR or immunostaining) are resulting in more-reliable detection of LCH cells in the bone marrow. High-risk patients are typically younger than 2 years. High-risk patients with intestinal involvement have a greater risk of not responding to therapy (49% do not respond to therapy) than patients without intestinal involvement (28% do not respond to therapy).[43] Nonetheless, intestinal disease is not an official criterion for high-risk disease.
- Low-risk organs include the skin, bone, lung, lymph nodes, gastrointestinal tract, pituitary gland, thyroid, thymus, and CNS. Involvement of every organ except kidney and gonads has been described.
Patients may present with single-organ involvement (single-system LCH), which may involve a single site (unifocal) or multiple sites (multifocal). Bone is the most common single-organ site. Less commonly, LCH may involve multiple organs (multisystem LCH), which may involve a limited number of organs, or it may be disseminated. Patients can have LCH of the skin, bone, lymph nodes, and pituitary gland in any combination and still be considered at low risk of death, although there may be a relatively high risk of developing long-term consequences of the disease.
Treatment decisions for patients are based on whether high-risk or low-risk organs are involved and whether LCH presents as unifocal, multifocal, or multisystem disease.
Single-system low-risk disease presentation
In single-system low-risk LCH, the disease presents with involvement of a single site or organ, including the following:
Bone
Bone is the most commonly affected system, estimated to be involved in 80% of patients with LCH. LCH can occur in any bone of the body, although the hands and feet are often spared.[44]
Sites of LCH bone lesions in children include the following:
- Lytic lesion of the skull: The most frequent site of LCH in children is a lytic lesion of the skull vault,[45] which may be asymptomatic or painful. It is often surrounded by a soft tissue mass that may extend internally to impinge on the dura. However, the presence of this mass does not affect prognosis.
- Femur, ribs, humerus, pelvis, and vertebra: Other frequently involved skeletal sites are femur, ribs, humerus, pelvis, and vertebra. Spine lesions may involve any vertebra, although involvement of the cervical vertebrae is most common, and spine lesions are frequently associated with other bone lesions. Spine lesions may result in collapse of the vertebral body (vertebra plana). Vertebral lesions with soft tissue extension often present with pain and may present with significant neurological deficits.[46] This finding is an indication for evaluating spinal cord compression with MRI scan.
- CNS-risk bones: Lesions of the facial bones or anterior or middle cranial fossae (e.g., temporal, orbit, sphenoid, ethmoid, zygomatic) with intracranial tumor extension comprise a CNS-risk group. These patients have a threefold increased risk of developing other CNS disease and diabetes insipidus. Systemic treatment is recommended for these patients because of the increased risk of diabetes insipidus. Proptosis from an LCH mass in the orbit mimics rhabdomyosarcoma, neuroblastoma, and benign fatty tumors of the eye.[47]
Skin and nails
-
Infants: Seborrheic involvement of the scalp may be mistaken for prolonged cradle cap in infants, unless the classic purpuric component is present. The second most common site involves the body creases, such as the antecubital fossa and perineum. Infants with LCH may also present with a generalized skin rash, which may mimic many other skin disorders and may or may not be pruritic. Vesicular LCH skin lesions need to be differentiated from congenital infections.
Skin LCH in infants may be limited to skin (skin-only disease) or may be part of multisystem LCH. In a report of 61 neonatal cases from 1,069 patients in the Histiocyte Society database, nearly 60% (36 of 61 patients) had multisystem disease, and 72% of the patients with multisystem disease had risk-organ involvement.[31] A retrospective analysis of 71 infants and children with apparent skin-only LCH found that those older than 18 months were more likely to have multisystem involvement and often relapsed after treatment with vinblastine and prednisone.[48] Eight of 11 patients in this category had circulating cells with the BRAF V600E variant, compared with only 1 of 13 patients in the skin-only group. Patients younger than 1 year with skin-only disease who were completely evaluated to exclude any other site of disease had a 3-year progression-free survival rate of 89% with initial therapy.
Skin-only LCH may be self-limited because the lesions may disappear without therapy during the first year of life. Therapy is used only for very extensive rashes, pain, ulceration, or bleeding. These patients must be monitored closely because skin-only LCH in neonates and very young infants may progress within weeks or months to high-risk multisystem disease, which may be life-threatening.[49,50,51]
In a review of patients presenting in the first 3 months of life with skin-only LCH, the clinical and histopathological findings of 21 children whose skin LCH regressed were compared with those of 10 children whose disease did not regress.[50] Patients with regressing disease had distal lesions that appeared in the first 3 months of life and were necrotic papules or hypopigmented macules. Patients with nonregressing disease who required systemic therapy more often had lesions in intertriginous areas. Immunohistochemical studies showed no difference in interleukin (IL)-10, Ki-67, E-cadherin expression, or T-reg number between the two clinical groups.
Hashimoto-Pritzker disease or congenital spontaneous regressing skin histiocytosis is a self-limited disease that has the same immunohistochemical staining as LCH but, on electron microscopy, shows dense bodies thought to be senescent mitochondria.[52] Careful review of the original cases revealed that some patients progressed to multisystem LCH; the distinction between skin-only LCH and Hashimoto-Pritzker disease is felt to be without clinical value because all of these infants should be carefully observed after diagnosis. It is not yet clear if the presence or absence of a BRAF V600E variant can be used to define whether systemic therapy is needed in skin-only LCH.
-
Children and adults: Children and adults may develop a red papular rash in the groin, abdomen, back, or chest that resembles a diffuse candidal rash. Seborrheic involvement of the scalp may be mistaken for a severe case of dandruff in older individuals. Ulcerative lesions behind the ears, involving the scalp, under the breasts, on the genitalia, or in the perianal region are often misdiagnosed as bacterial or fungal infections. Vesicular lesions may be seen and need to be differentiated from herpetic lesions.
Fingernail involvement is an unusual finding that may present as a single site or with other sites of LCH involvement. In this scenario, there are longitudinal, discolored grooves and loss of nail tissue. This condition often responds to the usual LCH therapies.[53]
Oral cavity
In the mouth, presenting symptoms include gingival hypertrophy and ulcers on the soft or hard palate, buccal mucosa, or tongue and lips. Hypermobile teeth (floating teeth) and tooth loss usually indicate involvement of underlying bone.[54,55] Lesions of the oral cavity may precede evidence of LCH elsewhere.
Lymph nodes and thymus
The cervical nodes are most frequently involved and may be soft-matted or hard-matted groups with accompanying lymphedema. An enlarged thymus or mediastinal node involvement can mimic an infectious process and may cause asthma-like symptoms. Accordingly, biopsy with culture is indicated for these presentations. Mediastinal involvement is rare (<5%) and usually presents with respiratory distress, superior vena cava syndrome, or cough and tachypnea. The 5-year survival rate for these patients is 87%, with deaths mostly attributable to hematologic involvement.[56]
Lung
In LCH, the lungs are less frequently involved in children than in adults because smoking in adults is a key etiologic factor.[57] Of 1,482 children in the French LCH registry, 7.4% of patients had pulmonary involvement and 1% of patients had severe disease requiring intensive care admission with multiple chest tube insertions for pneumothoraces and, sometimes, pleurodeses.[58] A review of 178 LCH cases from another center found that pulmonary involvement occurred in approximately 13 children (7.3%), 3 of whom had multisystem high-risk disease.[59] Multivariate analysis of pulmonary disease in multisystem LCH did not show pulmonary disease to be an independent prognostic factor. The 5-year OS rates were 94% for those with pulmonary involvement and 96% for those without pulmonary involvement.[26] Isolated pulmonary involvement is rarely seen in children.
The cystic/nodular pattern of disease reflects the cytokine-induced destruction of lung tissue. Classically, the disease is symmetrical and predominates in the upper and middle lung fields, sparing the costophrenic angle and giving a very characteristic picture on high-resolution CT scan.[60] Confluence of cysts may lead to bullous formation, and spontaneous pneumothorax can be the first sign of LCH in the lung, although patients may present with tachypnea or dyspnea. Ultimately, widespread fibrosis and destruction of lung tissue may lead to severe pulmonary insufficiency. Declining diffusion capacity may also indicate the onset of pulmonary hypertension.[39]
Widespread fibrosis and declining diffusion capacity are much less common in children. In young children with diffuse disease, therapy can halt the progress of the tissue destruction, and normal repair mechanisms may restore lung function, although scarring or even residual nonactive cysts may continue to be visible on radiological studies.
Pituitary gland
The posterior part of the pituitary gland and pituitary stalk can be affected in patients with LCH, causing central diabetes insipidus. Anterior pituitary involvement often results in growth failure and delayed or precocious puberty. Rarely, hypothalamic involvement may cause morbid obesity. For more information about diabetes insipidus, see the Endocrine system section.
Thyroid gland
Thyroid involvement has been reported in LCH. Symptoms include massive thyroid enlargement, hypothyroidism, and respiratory symptoms.[61]
Multisystem low-risk disease presentation
Bone and other organ systems
Patients with LCH may present with multiple bone lesions as the only organ involved (single-system multifocal bone) or with bone lesions and other organ systems involved (multisystem including bone). A Japanese LCH study (JLSG-02) included patients with single-system multifocal bone presentation and patients with multisystem-including-bone presentation. A review of the study found that patients in the multisystem-including-bone group were more likely to have lesions in the temporal bone, mastoid/petrous bone, orbit, and zygomatic bone (i.e., CNS-risk bones).[62] These patients also had a higher incidence of diabetes insipidus, correlating with the higher frequency of risk-bone lesions. A study from the Histiocyte Society found decreased mortality in patients with high-risk multisystem LCH who had bone involvement, suggesting that those with bone LCH may have more indolent disease.[63]
Abdominal organs and gastrointestinal system
In LCH, the liver and spleen are considered high-risk organs, and involvement of these organs affects prognosis. For more information, see the sections on Liver (sclerosing cholangitis) and Spleen.
Although rare, LCH infiltration of the pancreas and kidneys has been reported.[64]
Patients with diarrhea, hematochezia, perianal fistulas, or malabsorption have been reported.[65,66]
Endocrine system
Diabetes insipidus, caused by LCH-induced damage to the antidiuretic hormone-secreting cells of the posterior pituitary, is the most frequent endocrine manifestation in LCH.[67] MRI scans usually show nodularity and/or thickening of the pituitary stalk and loss of the pituitary bright spot on T2-weighted images. When the pituitary stalk is thickened or is very large, there is a 50% chance the patient will have a germinoma, LCH, or lymphoma.[68] Pituitary biopsies are rarely done. A biopsy of the pituitary gland may be indicated when the pituitary gland is the only site of disease and the stalk is thicker than 6.5 mm or there is a hypothalamic mass.[69] If the pituitary disease is associated with other sites of involvement, these other sites can be biopsied to establish the diagnosis.
Approximately 4% of patients with LCH present with an apparently idiopathic form of diabetes insipidus before other lesions of LCH are identified. A prospective follow-up study included pediatric patients who presented with idiopathic central diabetes insipidus and received only diabetes insipidus therapy. The study showed that 19% of patients eventually developed signs of LCH, while 18% were diagnosed with craniopharyngiomas and 10% with germinomas.[70] A prospective study of the etiology of central diabetes insipidus in children and young adults found that 15% of patients had LCH, 11% had germinomas, and 7% had craniopharyngiomas.[71] The other diagnoses were related to trauma, familial association, or midline defects, and 50% remained idiopathic. Decisions about whether or when to treat a patient with apparent isolated central diabetes insipidus as LCH without a biopsy remain controversial.
The approach is different for patients with known LCH and diabetes insipidus. These patients are 50% to 80% more likely to develop other lesions that are diagnostic of LCH (including bone, lung, and skin lesions) within 1 year of diabetes insipidus onset.[69,72] In general, patients with LCH present with diabetes insipidus later in the course of the disease, as noted in the following studies:
- One study compared the incidence of diabetes insipidus in patients who received no systemic therapy with that in patients who received 6 months of vinblastine/prednisone therapy. Patients who received no systemic therapy had a 40% incidence of diabetes insipidus. Patients who were treated with chemotherapy had a 20% incidence of diabetes insipidus. This finding strongly supports treatment of CNS-risk bone lesions, even when the disease is isolated to a single bony site.[73]
- In a study of 589 patients with LCH, the 10-year risk of pituitary involvement was 24%.[67] Diabetes insipidus was seen at a mean of 1 year after LCH diagnosis. Fifty-six percent of patients with LCH who developed diabetes insipidus developed anterior pituitary hormone deficiencies (growth, thyroid, or gonadal-stimulating hormones) within 10 years of the onset of diabetes insipidus. In this study, no decrease in the incidence of diabetes insipidus was seen in chemotherapy-treated patients, but this may reflect the length of the therapy and/or the number of drugs used.[67]
- Giving therapy for a longer duration and with more chemotherapeutic agents, the German-Austrian-Dutch (Deutsche Arbeitsgemeinschaft für Leukämieforschung und Behandlung im Kindesalter [DAL]) group found a cumulative incidence of diabetes insipidus of 20% at 15 years after LCH diagnosis.[73] The incidence of diabetes insipidus was also lower in patients treated with more-intensive chemotherapy regimens on the HISTSOC-LCH-III (NCT00276757), JLSG-96, and JLSG-02 studies in Japan (8.9% for multisystem patients) compared with the HISTSOC-LCH-I and HISTSOC-LCH-II studies (14.2%).[27,28,29,74,75] Overall, diabetes insipidus occurred in 11% of patients treated with multiagent chemotherapy and in up to 50% of patients treated less aggressively.[76,77]
Patients with multisystem disease and craniofacial involvement (particularly of the orbit, mastoid, and temporal bones) at the time of diagnosis carried a significantly increased risk of developing diabetes insipidus during the disease course (relative risk, 4.6). Of LCH patients with diabetes insipidus, 75% had these CNS-risk bone lesions.[73] The risk of diabetes insipidus increased when LCH remained active for a longer period of time or reactivated.
Approximately 50% of patients who present with isolated diabetes insipidus (as the initial manifestation of LCH) either have anterior pituitary deficits at the time of diagnosis or develop them within 10 years of diabetes insipidus onset.[72,77] Anterior pituitary deficits include secondary amenorrhea, panhypopituitarism, growth hormone deficiency, hypoadrenalism, and abnormalities of gonadotropins. The incidence of anterior pituitary deficits appears to be higher in patients with LCH than in those with true idiopathic central diabetes insipidus.
Ocular
Ocular LCH, although rare, has been reported and can sometimes lead to blindness. Other organ systems may be involved, and ocular LCH may not respond well to conventional chemotherapy.[47]
CNS
CNS disease manifestations
Patients with LCH may develop mass lesions in the hypothalamic-pituitary region, the choroid plexus, the grey matter, or the white matter.[78] These lesions contain CD1a-positive LCH cells and CD8-positive lymphocytes and are, therefore, active LCH lesions.[79]
Patients with large pituitary tumors (>6.5 mm) have a higher risk of anterior pituitary dysfunction and neurodegenerative CNS LCH.[80] A retrospective study of 22 patients found that all had radiological signs of neurodegenerative CNS LCH detected at a median time of 3 years and 4 months after LCH diagnosis; it worsened in 19 patients. Five patients had neurological dysfunction, 18 of 22 patients had anterior pituitary dysfunction, and 20 had diabetes insipidus. Growth hormone deficiency occurred in 21 patients. Luteinizing hormone/follicle-stimulating hormone deficiency occurred in 10 patients. Thyroid hormone deficiency occurred in 10 patients.
Clinical neurodegenerative syndrome LCH (cND-LCH)
A chronic neurodegenerative syndrome, cND-LCH, occurs in 1% to 4% of patients with LCH. These patients may develop tremors, gait disturbances, ataxia, dysarthria, headaches, visual disturbances, cognitive and behavioral problems, and psychosis.
Among 1,897 patients with LCH, 36 patients were diagnosed with cND-LCH. The incidence of cND-LCH was 4.1% at 10 years of follow-up. cND-LCH was more frequent in patients with pituitary involvement (86.1% vs. 12.2% without pituitary lesions), skin involvement (75% vs. 34.2% without skin lesions), and base skull bone involvement (63.9% vs. 28.4% without skull lesions). Patients with the BRAF variant were more likely to have cND-LCH (93.7%) than those without the variant (54.1%). In the multivariable analysis, the odds ratio of developing cND-LCH was 2.13 for patients with base skull lesions, 9.8 for patients with the BRAF V600E variant, and 30.88 for patients with pituitary involvement. The risk of cND-LCH had not plateaued up to 20 years after LCH diagnosis.[81]
Brain MRI scans from these patients show hyperintensity of the dentate nucleus and white matter of the cerebellum on T2-weighted images or hyperintense lesions of the basal ganglia on T1-weighted images and/or atrophy of the cerebellum.[25] The radiological findings may precede the onset of symptoms by many years or be found coincidently. One study included 83 patients with LCH who had at least two MRI studies of the brain for evaluation of craniofacial lesions, diabetes insipidus, and/or other endocrine deficiencies of neuropsychological symptoms.[36] Forty-seven of 83 patients (57%) had radiological neurodegenerative changes at a median time of 34 months from diagnosis of LCH. Of the 47 patients, 12 (25%) developed clinical neurological deficits that presented 3 to 15 years after the LCH diagnosis. Fourteen of the 47 patients had subtle deficits in short-term auditory memory.
The first histological evaluation of neurodegenerative lesions reported prominent T-cell infiltration, usually in the absence of the CD1a-positive dendritic cells, along with microglial activation and gliosis.[79] However, in a report from 2018, analysis of brain tissue from patients with neurodegenerative-disease LCH showed perivascular infiltration of CD207-negative cells staining with the BRAF V600E altered protein in the pons, cerebellum, and basal ganglia. These are areas identified by the characteristic abnormal MRI findings on T2 fluid-attenuated inversion recovery (FLAIR) images. Quantitative PCR analysis of these areas showed increased numbers of BRAF-altered cells and elevated expression of osteopontin. Brain tissue in these areas showed active demyelination, correlating with the radiological findings and clinical deficits.[82]
A study evaluated CNS-related permanent consequences (neuropsychologic deficits) in 14 of 25 patients with LCH who were monitored for a median of 10 years.[83] Seven of these patients had diabetes insipidus, and five patients had radiographic evidence of LCH CNS neurodegenerative changes.[83] Patients with craniofacial lesions had lower performance and verbal IQ scores than those with other LCH lesions.
Treatment of low-risk disease: single-system or multisystem LCH
Over many years, national and international study groups have defined risk-based therapy groups for allocation of LCH patients on the basis of mortality risk and risk of late effects of the disease.
Depending on the site and extent of disease, treatment of LCH may include observation (after biopsy or curettage), surgery, radiation therapy, or oral, topical, and intravenous medication. The recommended duration of therapy is 12 months for patients who require chemotherapy for single-system bone, skin, or lymph node involvement.
For patients with high-risk and low-risk multisystem disease, the reactivation rate after 6 months of therapy was as high as 50% on the HISTSOC-LCH-I and HISTSOC-LCH-II trials.[28,84] The German-Austrian-Dutch (Deutsche Arbeitsgemeinschaft für Leukämieforschung und Behandlung im Kindesalter [DAL]) group trials treated patients for 1 year and had fewer relapses (29%).[76,85] On the basis of these findings, the HISTSOC-LCH-III trial was designed to administer 12 months of chemotherapy for all high-risk multisystem patients and to randomly assign low-risk multisystem patients to either 6 months or 12 months of therapy. In patients with low-risk or high-risk disease who received 12 months of therapy, the reactivation rate was significantly reduced to approximately 30%.[29]
The standard treatment for LCH is based on data from international trials with large numbers of patients. However, some patients may have LCH involving only the skin, mouth, pituitary gland, or other sites not studied in these international trials. In these cases, therapy recommendations are based on case series that lack the evidence-based strength of the trials.
Clinical trials organized by the Histiocyte Society have been accruing patients on childhood treatment studies since the 1980s. Information about centers enrolling patients on these trials can be found on the ClinicalTrials.gov website.
Treatment options for patients with low-risk, single-system or multisystem disease depend on the site of involvement, as follows:
Isolated skin involvement
Treatment options for patients with isolated skin involvement include the following:
- Observation. Observation is recommended for all pediatric patients with asymptomatic skin-only LCH.[48]
- Therapy. Therapy is suggested only for patients with symptomatic disease, which includes extensive rashes, pain, ulceration, or bleeding.
Patients with skin-only involvement need to have a complete staging evaluation because 41% of these patients referred to one center had multisystem disease requiring treatment.[48] Careful clinical (but not radiological) follow-up of young infants with skin-only LCH is suggested because progression to high-risk multisystem disease is possible. Young children with skin-only LCH should be monitored periodically for many years because 1 of 19 children and 1 of 25 children in two series developed late diabetes insipidus.[31,49]
For patients who require therapy, treatment options for symptomatic isolated skin lesions include the following:
- Topical steroids. Medium- to high-potency steroids are effective, but recurrence after discontinuation is common.[50]
- Oral methotrexate. Oral methotrexate (20 mg/m2) weekly for 6 to 12 months.[86]
- Oral hydroxyurea. Oral hydroxyurea (20 mg/kg) daily for at least 12 months.[87]
- Oral thalidomide/lenalidomide.[88,89] Oral thalidomide 50 mg to 200 mg nightly.[88] Oral thalidomide/lenalidomide may be effective for both pediatric and adult patients.
- Topical nitrogen mustard. Topical application of nitrogen mustard can be effective for cutaneous LCH that is resistant to oral therapies, but not for disease involving large areas of skin.[90,91]
- Psoralen and long-wave ultraviolet A radiation (PUVA) or UVB. Psoralen and PUVA or UVB can be effective in skin LCH, but its use is limited by the potential for late skin cancers, especially in patients with light skin tones.[92,93]
- Chemotherapy. Systemic chemotherapy may be used in severe and symptomatic cases.
- Radiation therapy. Although external-beam radiation therapy has been used, it has not proven to be reliably effective and may have severe side effects.[94,95]
Skeletal involvement
Single skull lesions of the frontal, parietal, or occipital regions, or single lesions of any other bone
Treatment options for patients with single skull lesions of the frontal, parietal, or occipital regions, or single lesions of any other bone, include the following:
- Curettage. Curettage only is the recommended therapy, when possible, for isolated bone lesions. Curettage plus injection of methylprednisolone may also be used. LCH bone lesions do not need complete excision because this may increase healing time and the risk of long-term complications. Complete excision of skull lesions, which may require grafting, is not necessary.
- Low-dose radiation therapy. Local radiation therapy could be considered for an isolated lesion.[94,96][Level of evidence C3] Low-dose radiation therapy (7–10 Gy) is effective,[95,97] but its use is limited in pediatric patients to lesions that threaten organ function or are painful and not amenable to other therapies.[98,99]; [100][Level of evidence C1] In a single-institution study of 39 patients with LCH (age range, 1.5–67 years; 15 patients aged <18 years) who received radiation therapy to 46 lesions, there were no local recurrences in the 31 bony sites (median radiation therapy dose, 10.8 Gy; range, 7.5–24 Gy), and the freedom from local failure rate was 63% at 3 years in the 15 nonbone lesions (95% confidence interval, 32%–83%; P = .0008). In this study, no subsequent cancers occurred,[94] although subsequent cancers have been previously reported.[101] Skeletal complications are uncommon after the low doses that are used, but they can occur.[96]
Skull lesions in the mastoid, temporal, or orbital bones
The CNS-risk bones include the mastoid, temporal, spheroidal, zygomatic, ethmoidal, maxillary, orbital bones, sinuses, and lesions of the anterior or middle cranial fossa. Risk refers to the increased risk of progression to diabetes insipidus followed by brain (CNS) involvement.
The purpose of treating patients with isolated CNS-risk lesions is to decrease the chance of developing diabetes insipidus and other long-term neurological problems.[27]
Treatment options for patients with skull lesions in the mastoid, temporal, or orbital bones include the following:
-
Chemotherapy. The current treatment for CNS-risk bones is 12 months of vinblastine/prednisone therapy, as per the HISTSOC-LCH-III (NCT00276757) study:[27,29][Level of evidence B1]
- Weekly vinblastine (6 mg/m2) for 7 weeks for good response.
- Daily prednisone (40 mg/m2) for 4 weeks, then tapered over 2 weeks.
- Afterward, prednisone is given for 5 days at 40 mg/m2 every 3 weeks with the vinblastine injections (also every 3 weeks).
There is controversy about whether systemic therapy is required for the first presentation of unifocal bone LCH, even in the CNS-risk bones. One retrospective review reported a series of patients with orbital or mastoid lesions who underwent only surgical curettage. The treatment was completed by a single surgeon, specialized in orbital, ear, nose, or throat diseases.[102] None of these patients developed diabetes insipidus.
However, when comparing the incidence rates of diabetes insipidus in patients who received little or no chemotherapy (20%–50% incidence) with the incidence rates reported by the German-Austrian-Dutch group DAL-HX 83 trial (10% incidence in patients treated for LCH), it appears that the weight of evidence from the DAL-HX 83 trial supports chemotherapy treatment to prevent diabetes insipidus in patients with LCH in CNS-risk bones.[76,77] It should be noted, however, that the DAL-HX studies administered more drugs and treated patients for 12 months.
Vertebral or femoral bone lesions at risk of collapse
Treatment options for patients with vertebral or femoral bone lesions at risk of collapse include the following:
- Observation. A single vertebral body lesion without soft tissue extension to the extradural space may be observed only.[103]
- Low-dose radiation therapy. Low-dose radiation therapy may be used to promote resolution in an isolated vertebral body lesion or a large femoral neck lesion at risk of fracture, where chemotherapy is not usually indicated (single bone lesion). Despite the low dose required (7–10 Gy), radiation therapy should be used with caution because of concerns about secondary malignancies in adjacent tissues, skeletal deformities if the growth plates are irradiated in very young children,[96,101] or if the thyroid gland would be in the radiation field in cervical vertebral lesions.
- Chemotherapy. Patients with soft tissue extension from vertebral lesions are often treated successfully with chemotherapy,[46][Level of evidence C2] but prolonged therapy does not appear to be needed beyond the period required to reduce the mass and any risk to the spinal cord. The risk of reactivation of a single bone lesion was only 9% in one large retrospective series.[104]
- Bracing or spinal fusion. When instability of the cervical vertebrae and/or neurological symptoms are present, bracing—or rarely, spinal fusion—may be needed.[105]
Multiple bone lesions (single-system multifocal bone lesions)
Treatment options for patients with multiple bone lesions (single-system multifocal bone lesions) at risk of collapse include the following:
-
Chemotherapy. The most commonly used systemic chemotherapy regimen is the combination of vinblastine and prednisone. Based on the results of the HISTSOC-LCH-III (NCT00276757) trial, 12 months of treatment with weekly vinblastine (6 mg/m2) for 7 weeks, then every 3 weeks, is used for good responders.[29] Prednisone (40 mg/m2) is given daily for 4 weeks, then tapered over 2 weeks. Afterward, prednisone is given for 5 days at 40 mg/m2 every 3 weeks with the vinblastine injections.
A short treatment course (<6 months) with only a single agent (e.g., prednisone) is not sufficient, and the number of relapses is higher. A reactivation rate of 18% was reported with a multidrug treatment regimen that was used for 6 months versus a historical reactivation rate of 50% to 80% with surgery alone or with a single-drug treatment regimen.[106] A comparison of results from two trials in Japan revealed no improvement in progression-free survival rates (66% vs. 65%) when additional prednisone and a prolonged maintenance phase were added.[107]
For information about additional agents used to treat multifocal bone LCH, see the Multiple bone lesions in combination with skin, lymph node, or diabetes insipidus (low-risk multisystem LCH) section.
Multiple bone lesions in combination with skin, lymph node, or diabetes insipidus (low-risk multisystem LCH)
Treatment options for patients with multiple bone lesions in combination with skin, lymph node, or diabetes insipidus (low-risk multisystem LCH) include the following:
-
Chemotherapy (vinblastine and prednisone in combination). Based on the results of the randomized HISTSOC-LCH-III (NCT00276757) trial, the same chemotherapy regimen of vinblastine and prednisone, as described above, is used for 12 months. Patients without risk-organ involvement who were randomly assigned to receive 12 months of treatment with vinblastine/prednisone had a lower 5-year reactivation rate (37%) than did patients who received only 6 months of treatment (54%; P = .03) and patients treated with historical 6-month schedules (52% [HISTSOC-LCH-I] and 48% [HISTSOC-LCH-II]; P < .001). Most disease reactivations were in bone, skin, or other non-risk locations.[29]
Patients with low-risk multisystem LCH have a survival rate of almost 100%, but reactivations were shown to be major risk factors for significant late effects on the DAL and Histiocyte Society trials.[29,76]
-
Chemotherapy (other regimens). Other chemotherapy regimens have also been effective, including the following:
- Vincristine, cytarabine, and prednisone in combination.[108][Level of evidence C2] This combination has proven effectiveness as frontline or salvage therapy. However, prednisone is now given for a much shorter time than was originally published (52 weeks): 4 weeks at 40 mg/m2 then tapered to 20 mg/m2 by week 6 during the induction phase, and for 5 days every 3 weeks at 20 mg/m2 with a single dose of vincristine and 5 days of cytarabine during maintenance.
- Cladribine. Cladribine given at 5 mg/m2 per day for 5 days every 3 weeks for two to six cycles can be an effective salvage therapy for recurrent bone or low-risk multisystem disease.[109][Level of evidence C2] More than six cycles is not recommended because of the risk of cumulative cytopenias.
-
Bisphosphonate therapy. Bisphosphonate therapy can also be effective for treating LCH bone lesions.[110,111][Level of evidence C2] A nationwide survey from Japan described 16 children treated with bisphosphonates for bone LCH. None had risk-organ disease. Most patients received six cycles of pamidronate at 1 mg/kg per course given at 4-week intervals. In 12 of 16 patients, all active lesions including skin (n = 3) and soft tissues (n = 3) resolved. Eight patients remained disease free at a median of 3.3 years.[112] Other bisphosphonates such as zoledronate have been used to successfully treat bone LCH.[113]
Although bisphosphonates are used for bone LCH, some publications report response in other organs, such as skin.[111,112]
CNS disease
CNS lesions
CNS LCH lesions include the following:
- Mass lesions or tumors in the cerebrum, cerebellum, or choroid plexus.
- Mass lesions of the hypothalamic-pituitary axis that are always associated with diabetes insipidus and are often associated with other endocrinopathies.
Drugs that cross the blood-brain barrier, such as cladribine, or other nucleoside analogs, such as cytarabine, are used for active CNS LCH lesions.
Treatment options for patients with CNS LCH lesions include the following:
- Chemotherapy (cladribine). Treatment of mass lesions with cladribine has been effective in 13 reported cases.[114,115]; [116][Level of evidence C2] Mass lesions included enlargement of the hypothalamic-pituitary axis, parenchymal mass lesions, and leptomeningeal involvement. Doses of cladribine ranged from 5 mg/m2 to 13 mg/m2, given at varying frequencies.[116][Level of evidence C2]
- Chemotherapy (other regimens). Patients with LCH and mass lesions in the hypothalamic-pituitary region, the choroid plexus, the grey matter, or the white matter may also respond to standard LCH chemotherapy.[117,118][Level of evidence C3] Treatment with vinblastine with or without corticosteroids for patients with CNS mass lesions (20 patients; mainly pituitary) demonstrated objective responses in 15 patients. Of 20 patients, 5 achieved complete responses and 10 achieved partial responses.
Clinical neurodegenerative syndrome LCH (cND-LCH)
There is no established optimal therapy for cND-LCH, and assessment of response can be difficult.[119]
In cND-LCH, T2 FLAIR hyperintense signals are present, most often in the cerebellar white matter, pons, basal ganglia, and, sometimes, in the cerebrum. It is not clear whether LCH changes in the cerebellum, pons, and basal ganglia diagnosed by MRI and without clinical neurological findings should be treated. Early studies suggested that not all LCH-related radiological changes progressed to clinical neurodegenerative disease. However, treatment in the early stages of clinical disease before permanent damage occurs appears to be important. The current recommendation is ongoing neurological evaluation both clinically and with MRI scanning. Therapy starts as soon as clinical neurodegenerative disease progression is noted. It is unclear whether progressive radiological changes should be an indication for treatment.[38]
Other drugs used in active LCH, such as dexamethasone, cladribine, and infliximab, have been used in small numbers of patients with mixed results. Many of these agents may result in the complete or partial resolution of radiographic findings, but definitive clinical response rates have not been rigorously defined.[38,120,121,122,123]; [116][Level of evidence C2]
Newer treatment options for patients with cND-LCH include the following:
-
BRAF V600E inhibitor therapy. For more information, see the Treatment of recurrent, refractory, or progressive high-risk disease: multisystem LCH section.
Clinical experience suggests that BRAF V600E inhibitor therapy may be the most effective therapy for improving neurological symptoms in cND-LCH, but the therapy may need to be continued lifelong.[82][Level of evidence C3]; [124]
-
Chemotherapy. A study using cytarabine with or without vincristine for up to 24 months reported improved clinical and MRI findings in some patients and stable disease in the others.[38][Level of evidence C1] Seven of eight patients were monitored for more than 8 years after stopping therapy and had stable neurological and radiographic findings.
In the Japan LCH Study Group (JLSG)-96 Protocol, cytarabine failed to prevent the onset of neurodegenerative syndrome. Patients received cytarabine 100 mg/m2 daily on days 1 to 5 during induction and 150 mg/m2 on day 1 of each maintenance cycle (every 2 weeks for 6 months). Three of 91 patients developed neurodegenerative disease, which is similar to the rate reported for patients on the Histiocyte Society studies.[125][Level of evidence B4]
- Rituximab. Eight patients with neurological symptoms for a median of 8 years and who developed new symptoms after being treated with cytarabine received rituximab (375 mg/m2 weekly for 4 weeks, repeated every 3 months and increased to 555 mg/m2 for no improvement or worsening of symptoms) for variable lengths of time. Clinical symptoms improved in seven of eight patients (five patients improved within 1 month of starting rituximab). Five patients remained free of progressive clinical symptoms for 3 years or longer.[126][Level of evidence C3]
Early recognition of clinical neurodegeneration and early institution of therapy appear to be vital for success of therapy. Studies combining MRI findings together with CSF markers of demyelination, to identify patients who require therapy even before onset of clinical symptoms, are under way in several countries. Studies of CSF and serum biomarkers in an attempt to predict and prevent neurodegenerative disease are also ongoing.[119]
High-Risk Disease: Multisystem LCH
Clinical presentation of high-risk multisystem LCH
Liver (sclerosing cholangitis)
The liver may be enlarged from direct infiltration of LCH cells or as a secondary phenomenon of excess cytokines, which cause macrophage activation or infiltration of lymphocytes around bile ducts. LCH cells have a portal (bile duct) tropism that may lead to biliary damage and ductal sclerosis. Peribiliary LCH cells and, rarely, nodular masses of LCH may also be present.[127]
Sonography, CT, or MRI of the liver will show hypoechoic or low-signal intensity along the portal veins or biliary tracts when the liver is involved with LCH.[127] While ultrasonography and/or MRI-cholangiogram can be helpful in the diagnosis of this complication, liver biopsy is the only definitive way to determine whether active LCH or residual hepatic fibrosis is present. Biopsy results often show lymphocytes and biliary obstructive effects without LCH cells.[128]
Patients with hepatic LCH present with hepatomegaly (>3 cm below the costal margin in the midclavicular line) or hepatosplenomegaly and dysfunction, as evidenced by hypoproteinemia (<55 g/L, hypoalbuminemia <25 g/L), or histological findings of active disease.[29] Patients may also have elevated alkaline phosphatase, liver transaminases, and gamma glutamyl transpeptidase levels, clotting dysfunction, or present with ascites.
One of the most serious complications of hepatic LCH is cholestasis and sclerosing cholangitis.[129] This usually occurs months after initial presentation, but occasionally may be present at diagnosis. The median age of children with this form of hepatic LCH is 23 months. The natural history of sclerosing cholangitis is variable. Some patients who are treated with chemotherapy improve, while other patients have stable disease or progress from sclerosis to biliary cirrhosis and portal hypertension, which may be seen even in the absence of active LCH cells. A report of 13 patients with LCH and liver disease found that all patients had BRAF V600E variants in skin, bone, or liver biopsy samples.[130] It is not known whether the early use of inhibitor therapy in this group of patients will reduce or prevent the progression of sclerosing cholangitis. This therapy remains to be investigated.
Spleen
Massive splenomegaly (usually >2 cm below costal margin in the midclavicular line),[29] resulting from either primary involvement by LCH or from portal hypertension secondary to biliary cirrhosis, may lead to cytopenias because of hypersplenism and may cause respiratory compromise. Splenectomy typically provides only transient relief of cytopenias, as increased liver size and reticuloendothelial activation result in peripheral blood cell sequestration and destruction. Splenectomy is performed only as a life-saving measure.
Bone marrow
Most patients with bone marrow involvement are young children who have diffuse disease in the liver, spleen, lymph nodes, and skin and who present with significant thrombocytopenia (<100,000 × 109 /L) and anemia (hemoglobin <10 g/dL; infants, <9 g/dL) not secondary to other causes, with or without leucopenia (<4.0 × 109 /L).[29,131] Other patients have only mild cytopenias and are found to have bone marrow involvement with LCH by sensitive immunohistochemistry, flow cytometry, or PCR for analysis of BRAF-altered cells in the bone marrow.[132,133] A large number of macrophages can obscure LCH cells in the bone marrow.[134] Patients with LCH who are considered at very high risk sometimes present with hemophagocytosis in the bone marrow.[135] The cytokine milieu driving LCH is probably responsible for the epiphenomenon of macrophage activation which, in the most severe cases, presents with typical manifestations of hemophagocytic lymphohistiocytosis such as cytopenias and hyperferritinemia.
Treatment of high-risk multisystem LCH
Over many years, national and international study groups have defined risk-based therapy groups for allocation of LCH patients on the basis of mortality risk and risk of late effects of the disease.
Depending on the site and extent of disease, treatment of LCH may include observation (after biopsy or curettage), surgery, radiation therapy, or oral, topical, and intravenous medication. The recommended duration of therapy is 12 months for patients who require chemotherapy for single-system bone, skin, or lymph node involvement.
For patients with high-risk and low-risk multisystem disease, the reactivation rate after 6 months of therapy was as high as 50% on the HISTSOC-LCH-I and HISTSOC-LCH-II trials.[28,85] The German-Austrian-Dutch (DAL) group trials treated patients for 1 year and had fewer relapses (29%).[76,85] On the basis of these findings, the HISTSOC-LCH-III trial was designed to administer 12 months of chemotherapy for all high-risk multisystem patients and to randomly assign low-risk multisystem patients to either 6 months or 12 months of therapy. In patients with low-risk or high-risk disease who received 12 months of therapy, the reactivation rate was significantly reduced to approximately 30%.[29]
The standard treatment for LCH is based on data from international trials with large numbers of patients. However, some patients may have LCH involving only the skin, mouth, pituitary gland, or other sites not studied in these international trials. In these cases, therapy recommendations are based on case series that lack the evidence-based strength of the trials.
Clinical trials organized by the Histiocyte Society have been accruing patients on childhood treatment studies since the 1980s. Information about centers enrolling patients on these trials can be found on the ClinicalTrials.gov website.
Treatment options for patients with high-risk multisystem disease (spleen, liver, and bone marrow involving one or more sites) include the following:
Chemotherapy
Evidence (chemotherapy):
- In the HISTSOC-LCH-II and HISTSOC-LCH-III (NCT00276757) studies, the standard treatment arm consisted of vinblastine and prednisone, as described above, but mercaptopurine was added to the continuation phase of the protocol.[27,76][Level of evidence A1]
- The standard therapy length recommended for LCH involving the spleen, liver, or bone marrow (high-risk organs) is now 12 months, based on the DAL-HX 83 and HISTSOC-LCH-III studies.
- In the HISTSOC-LCH-II study, patients were randomly assigned to treatment with either vinblastine, prednisone, and mercaptopurine or vinblastine, prednisone, mercaptopurine, and etoposide.[28][Level of evidence A1]
- There was no statistically significant difference in outcomes (response at 6 weeks, 5-year probability of survival, relapses, and permanent consequences) between the two treatment groups. Hence, etoposide has not been used in subsequent Histiocyte Society trials.
- Late review of the results, however, reported reduced mortality for patients with risk-organ involvement in the etoposide arm.
- Although controversial, a comparison of patients in the HISTSOC-LCH-I trial with patients in the HISTSOC-LCH-II trial suggested the following results:[26]
- Increased treatment intensity promoted additional early responses and reduced mortality.
- It is important to note that those studies included lungs as risk organs. However, subsequent analyses have shown that lung involvement lacks prognostic significance.
- In the HISTSOC-LCH-III (NCT00276757) study, risk-organ–affected patients were randomly assigned to receive either vinblastine/prednisone/mercaptopurine or vinblastine/prednisone/mercaptopurine plus methotrexate (intravenous during the induction phase and oral in the continuation phase).[29]
- The response rates at 6 and 12 weeks and OS were no different between arms; however, there were significantly increased grade 3 and grade 4 toxicities in patients who received methotrexate.
- An important finding of the HISTSOC-LCH-III study was that the survival of patients with high-risk LCH on both arms of the study was significantly improved compared with that of patients on the earlier HISTSOC-LCH-II study, even though the standard arm used the same drugs. Possible explanations for better survival include the following:
- A second 6-week induction phase of weekly vinblastine with oral prednisone was administered for 3 days per week. This reinduction phase was given to all patients who did not achieve an NAD status by the end of the 6-week induction phase, before going onto the every-3-weeks maintenance courses. The rate of NAD increased after the second induction phase; this course may have played a significant role in the improved survival rate.
- Better supportive care.
- Earlier change to an effective salvage strategy for nonresponsive lesions.
- It should be noted that although survival was improved in the HISTSOC-LCH-III study, only 60% of patients achieved an NAD status in risk organs after a year of therapy, and 25% to 29% of patients relapsed.
- In the JLSG-96 trial, treatment included a 6-week induction regimen of cytarabine, vincristine, and prednisolone followed by 6 months of maintenance therapy with cytarabine, vincristine, prednisolone, and low-dose intravenous methotrexate. If patients had a poor response to the initial regimen, they were switched to a salvage regimen of intensive combination doxorubicin, cyclophosphamide, methotrexate, vincristine, and prednisolone.[125][Level of evidence B1]
- The 5-year response rate was 78%, and the OS rate was 95% for patients with multisystem disease.
- Diabetes insipidus occurred in 8.9% of patients with multisystem disease.
- Similar to the HISTSOC-LCH-III (NCT00276757) study, the important finding of this study was the increased survival compared with previous JLSG studies and the HISTSOC-LCH-II study. This was attributed to the early change to a more effective salvage therapy for patients with nonresponsive disease, as well as better supportive care.
- The study had a high reactivation rate, which prompted several changes, including an increase in the duration of the trial to 12 months and the addition of vinblastine, prednisone, mercaptopurine, and methotrexate.[62]
- The JLSG-02 protocol was similar to the JLSG-96 study, except that cyclosporine was added to the reinduction of poor responders and the length of treatment was increased to 54 weeks for good responders and 60 weeks for poor responders.[136][Level of evidence B1]
- Despite a markedly increased intensity of treatment, the event-free survival (EFS) rates were only 46% for high-risk patients and 70% for low-risk patients. In the HISTSOC-LCH-III study, the EFS rates were 33% for high-risk patients and 50% for low-risk patients.[137]
Treatment options under clinical evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
-
HISTSOC-LCH-IV (NCT02205762) (LCH-IV, International Collaborative Treatment Protocol for Children and Adolescents With LCH): On the basis of features at presentation and response to treatment, the LCH-IV study tailors treatment to one of the following seven strata:
- Stratum I: First-line treatment for multisystem LCH patients (group 1) and patients with single-system LCH with multifocal bone or CNS-risk lesions (group 2).
- Stratum II: Second-line treatment for non–risk-organ patients (patients without risk-organ involvement who fail first-line therapy or have a reactivation after completion of first-line therapy).
- Stratum III: Salvage treatment for risk-organ LCH (patients with dysfunction of risk organs who fail first-line therapy).
- Stratum IV: Hematopoietic stem cell transplant for risk-organ LCH (patients with dysfunction of risk organs who fail first-line therapy).
- Stratum V: Monitoring and treatment of isolated tumorous and neurodegenerative CNS LCH.
- Stratum VI: Natural history and management of other single-system LCH (patients who do not need systemic therapy at the time of diagnosis).
- Stratum VII (long-term follow up): All patients, regardless of previous therapy, will be monitored for reactivation or permanent consequences once complete disease resolution has been achieved and the respective protocol treatment has been completed.
- NCT02670707 (Cytarabine or Vinblastine Sulfate and Prednisone in Treating Patients With LCH): The purpose of this trial is to compare previously used vinblastine/prednisone to single therapy with cytarabine for LCH.
It is preferable that patients with LCH be enrolled in a clinical trial whenever possible so that advances in therapy can be achieved more quickly, using evidence-based recommendations, and to ensure optimal care. Information about clinical trials for LCH in children is available from the NCI website, Histiocyte Society website, and the North American Consortium for Histiocytosis (NACHO) website.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
Recurrent, Refractory, or Progressive Childhood LCH
Reactivation of single-system and multisystem LCH
Reactivation of LCH after complete response is common.[138] In a large study, the percentage of patients with reactivations was 9% to 17.4% for single-site disease; 37% for single-system, multifocal disease; 46% for multisystem (non–risk-organ) disease; and 54% for risk-organ involvement. Forty-three percent of reactivations were in bone, 11% in ears, 9% in skin, and 7% developed diabetes insipidus; a lower percentage of patients had lymph node, bone marrow, or risk-organ relapses.[138] The median time to reactivation was 12 to 15 months in non-risk patients and 9 months in high-risk patients. One-third of patients had more than one reactivation, varying from 9 to 14 months after the initial reactivation. Patients with reactivations were more likely to have long-term sequelae in the bones, diabetes insipidus, or other endocrine, ear, or lung problems.[138]
A comprehensive review of the German-Austrian-Dutch (DAL) and Histiocyte Society clinical trials revealed a reactivation rate of 46% at 5 years for patients with multisystem LCH, with most reactivations occurring within 2 years of first remission. A second reactivation occurred in 44% of patients, again within 2 years of the second remission. Involvement of the risk organs in these reactivations occurred only in those who were initially in the high-risk group (meaning they had liver, spleen, or bone marrow involvement at the time of original diagnosis).[84][Level of evidence C2] Most reactivations, even in patients with high-risk disease who initially responded to therapy, were in bone, skin, or other low-risk locations.
Consistent with these findings, the percentage of reactivations in multisystem disease was 45% in one trial from Japan [125][Level of evidence A1] and 46% in the HISTSOC-LCH-II trial.[28] There was no statistically significant difference in reactivations between the high-risk and low-risk groups. The DAL-HX studies and the studies from Japan concluded that intensified treatment increased the rapidity of response, particularly in young children and infants younger than 2 years, and together with rapid switch to salvage therapy for nonresponders, mortality was reduced for patients with high-risk multisystem LCH. Based on the HISTSOC-LCH-III (NCT00276757) randomized trial, prolongation of therapy also significantly reduced the rate of reactivation. The optimal duration of therapy (12 vs. 24 months) is being addressed in the HISTSOC-LCH-IV (NCT02205762) trial.
Treatment of recurrent, refractory, or progressive low-risk disease: single-system or multisystem LCH
The optimal therapy for patients with recurrent, refractory, or progressive LCH has not been determined.
Treatment options for patients with recurrent, refractory, or progressive low-risk, single-system or multisystem LCH include the following:
Chemotherapy
The following chemotherapy regimens have been used to treat patients with recurrent, refractory, or progressive low-risk disease:
- Vinblastine and prednisone. Patients with recurrent bone disease that recurs months after vinblastine and prednisone are stopped can benefit from treatment with a reinduction of vinblastine weekly and daily prednisone for 6 weeks. If there is NAD or very little evidence of active disease, treatment can be changed to every 3 weeks, with the addition of oral mercaptopurine nightly.[106]
- Vincristine, prednisone, and cytarabine. An alternative treatment regimen for patients with any combination of low-risk disease sites employs vincristine, prednisone, and cytarabine.[108][Level of evidence C3]
- Single-agent cytarabine. Single-agent cytarabine at doses of 100 to 170 mg/m2 per day for five days has also proven to be effective.[139]
- Cladribine. Cladribine at 5 mg/m2 per day for 5 days per course has demonstrated effectiveness for recurrent low-risk LCH (multifocal bone and low-risk multisystem LCH), with very little toxicity.[109][Level of evidence C3] Cladribine therapy should, if possible, be limited to a maximum of six cycles to avoid cumulative and potentially long-lasting cytopenias.
In a study of 44 pediatric patients with low-risk LCH who were treated with cladribine, 5 patients achieved complete remissions after a median follow-up of over 5 years.[140] Grade 3 or higher neutropenia occurred in 32% of patients, and grade 3 or higher lymphopenia occurred in 72% of patients. Patients with stable disease or partial responses after 6 months of treatment may ultimately attain a complete response.
- Clofarabine. Clofarabine is a proven effective therapy for patients with multiple relapses of low-risk or high-risk LCH.[141][Level of evidence C3]
- Hydroxyurea, alone or in combination with oral methotrexate. In one single-center trial, treatment with hydroxyurea, alone or in combination with oral methotrexate, reported the following results:[87][Level of evidence C3]
- Twelve of fifteen patients with low-risk recurrent LCH had responses to treatment.
- Thalidomide. A phase II trial of thalidomide in patients with LCH (ten low-risk patients; six high-risk patients) who failed primary treatment and at least one secondary regimen demonstrated the following:[88][Level of evidence C3]
- Of the ten low-risk patients, four had complete responses and three had partial responses. Complete response was defined as healing of bone lesions on plain radiographs (n = 3) or complete resolution of skin rash (n = 4, including 3 with bone lesions that had complete resolution). Partial response was defined as healing of a bone lesion, but then worsening of a skin rash that was partially resolved.
- Dose-limiting toxicities, such as neuropathy and neutropenia, may limit the overall usefulness of thalidomide.
- Thalidomide is not a significant agent in treating pediatric patients.
Bisphosphonate therapy
Bisphosphonate therapy is also effective for treating patients with recurrent LCH bone lesions.[142]
Evidence (bisphosphonate therapy):
- In a survey from Japan, 16 patients with bone lesions were treated with bisphosphonate therapy. None of the patients had risk-organ disease. Most patients received six cycles of pamidronate at 1 mg/kg per course, given at 4-week intervals.[110][Level of evidence C3]
- Of the 16 patients, 12 were successfully treated.
- Skin and soft tissue LCH lesions also resolved in the responding patients.
- Eight of the 12 patients remained disease free at a median of 3.3 years.
- Other bisphosphonates, such as zoledronate and oral alendronate, have also been successful in treating bone LCH.[111,112,113][Level of evidence C3]
Treatment of recurrent, refractory, or progressive high-risk disease: multisystem LCH
Data from the DAL group studies showed that patients with high-risk multisystem LCH who had progressive disease by week 6 of standard induction treatment or who did not achieve at least a partial response by week 12 had only a 10% chance of survival.[27] These results were consistent with those of the less-intensive HISTSOC-LCH-II trial in which patients treated with vinblastine/prednisone who did not respond well by week 6 had a 27% chance of survival, compared with 52% for good responders.[28][Level of evidence A1] To improve on these results, patients with poorly responsive disease need to move to salvage strategies by week 6 for progressive disease and no later than week 12 for those without at least a good response.
Treatment options for patients with recurrent, refractory, or progressive high-risk multisystem LCH include the following:
- Chemotherapy.
- Targeted therapy (e.g., MAPK inhibitors).
- Hematopoietic stem cell transplant (HSCT).
Chemotherapy
Cladribine and cytarabine
Evidence (cladribine and cytarabine):
- Ten patients with refractory high–risk-organ involvement (liver, spleen, or bone marrow) and resistant multisystem low–risk-organ involvement were treated with an intensive acute myeloid leukemia–like protocol consisting of cladribine and cytarabine.[143][Level of evidence C3] The follow-up HISTSOC-LCH-S-2005 trial accrued 27 patients and demonstrated the following results:[144]
- The progression-free survival rate was 63%, and the 5-year OS rate was 85% in this refractory, high-risk patient population.
- All patients developed grade 4 hematologic toxicity, and five of these patients had severe sepsis.
- For centers that cannot provide the intensive supportive care needed for this protocol, an alternative protocol using lower doses of cladribine (5 mg/m2 /day × 5 days) and cytarabine (100 mg/m2 /day × 4 days) was published.[145][Level of evidence C2]
- Six of nine patients achieved NAD status, and one patient had improved status after six courses.
- Some patients received maintenance therapy.
- Seven of nine patients remained in complete remission, with a median follow-up of 6.5 years.
Clofarabine
Patients who did not respond to treatment with cladribine were reported to respond to treatment with clofarabine.[146]; [147][Level of evidence C2]
Evidence (clofarabine):
- Eleven patients with recurrent multisystem high-risk and low-risk disease were treated with clofarabine.[141]
- The OS rate was 90%.
- If confirmed in prospective trials, the reduced toxicity of this regimen compared with the cladribine/cytarabine combination could be advantageous, despite the cost of the drug.
Targeted therapy
MAPK inhibitors
The discovery that most patients with LCH have BRAF V600E or other variants that result in activation of the RAS pathway suggests that new therapies that target molecules within this pathway (MAP2K/ERK inhibitors) will become an important part of LCH therapy.
Evidence (vemurafenib):
- Forty-four LCH patients with risk-organ involvement and ten LCH patients without risk-organ involvement were treated with vemurafenib. Of the 44 risk-organ–involved patients, 31 received vemurafenib as their original therapy and 13 received vemurafenib as treatment after disease progression. The ten risk-organ–negative patients also received vemurafenib after disease progression.[148][Level of evidence C3]
- After 8 weeks of treatment, there were 38 complete responses and 16 partial responses. Most patients were treated for 6 months.
- Thirty patients stopped taking vemurafenib; 24 of these patients subsequently relapsed: 72% of patients at 6 months and 84% of patients at 12 months off therapy.
- The relapse rate was 95% for patients with risk-organ involvement and 57% for patients without risk-organ involvement.
- Relapse was associated with the persistence of circulating BRAF-positive cells.
- The most frequent adverse effects of the drug were dermatologic. In a review of 57 patients with LCH who received vemurafenib for refractory disease, 72% of patients had cutaneous adverse events, 86% of which were grade 1 or grade 2.[149] Most patients had photosensitivity, keratosis pilaris, macular or follicular rashes, or xerosis. No skin tumors were observed.
- A systematic review and meta-analysis evaluated the efficacy and safety of vemurafenib for the treatment of patients with LCH. The analysis found 416 studies, 22 of which fit the inclusion criteria. There were 104 patients with relapsed or refractory disease and 3 patients with newly diagnosed disease.[150]
- With vemurafenib treatment, the median time to first response was 1 week and the median time to best response was 5.25 months.
- Sixty-two patients (58%) achieved NAD status, and 36% had decreased active disease.
- The overall response rate was 94.4%.
- Major toxicity included rash and photosensitivity.
- The authors concluded that vemurafenib was highly efficacious and safe to treat patients with refractory LCH, but the duration of therapy has yet to be established.
- A multicenter, retrospective analysis of experiences that used various MAPK inhibitors to treat 21 pediatric patients with LCH who had failed at least one previous therapy (median, three previous therapies) demonstrated the following:[124][Level of evidence C3]
- An overall response rate of 86% (complete response, 19%; partial response, 67%).
- Stable disease in 10% of patients.
- The most frequent toxicities were skin rashes and arthralgias. Other toxicities included neutropenia, fatigue, and uveitis.
Evidence (dabrafenib with or without trametinib):
- One study compared dabrafenib alone (13 patients) with dabrafenib and trametinib (12 patients) for the treatment of patients with relapsed or progressive LCH.[151]
- With a 2-year follow-up, the responses were similar in both arms (46.2% complete response, 30.8% regressive disease, and 23.1% stable disease for dabrafenib alone vs. 33.3% complete response, 25.0% regressive disease, and 25.0% stable disease for combination therapy).
- Adverse events were also similar and included pyrexia and vomiting, cough, and increased serum creatinine.
- This study would suggest that combination therapy is not more effective in patients with LCH. However, more data are needed.
Although malignancies such as squamous cell carcinoma have been reported in adults treated with MAPK inhibitors, such malignancies have not been reported in pediatric patients.[149] Like adults, children develop acneform rashes, photosensitivity, diarrhea, and, sometimes, myalgias.[124]
Tyrosine kinase inhibitors
Evidence (tyrosine kinase inhibitors):
- Imatinib has been shown to decrease differentiation of CD34-positive stem cells to dendritic cells. Small case reports of its efficacy in patients with LCH have been published.[152,153]
Hematopoietic stem cell transplant (HSCT)
HSCT has been used in patients with multisystem high–risk-organ involvement that is refractory to chemotherapy.[142,154,155,156,157] Early results showing very high treatment-related mortality in these ill young infants led to the development of reduced-intensity conditioning.
Evidence (reduced-intensity conditioning vs. myeloablative conditioning for HSCT):
- A review from the United Kingdom suggested that in transplant centers that have LCH HSCT experience, there was no advantage to reduced-intensity conditioning in their setting.[158][Level of evidence C2]
- Reduced-intensity conditioning provided no OS advantage over myeloablative conditioning for LCH patients; the relapse rate after reduced-intensity conditioning was significantly higher (28%) than the relapse rate after myeloablative conditioning (8%).
- Many of the patients who received reduced-intensity conditioning and relapsed were successfully re-treated with chemotherapy alone.
Treatment options for sclerosing cholangitis and macrophage activation
Seventy-five percent of children with sclerosing cholangitis will not respond to chemotherapy because the LCH is no longer active, but the fibrosis and sclerosis remain. Despite the limitations, liver biopsy may be the only way to distinguish active LCH from end-stage fibrosis. Liver transplant is the only alternate treatment when hepatic function worsens. A review of 60 patients with LCH (55 children) who underwent hepatic transplant for LCH-associated liver failure reported a 5-year survival rate of 82%. Posttransplant rejection occurred in 55% of patients, 22% of whom received a second transplant. The 5-year overall graft survival rate was 62% for patients who underwent deceased-donor liver transplant and 81% for patients who underwent living-donor liver transplant (not statistically significant). Nine patients died (15%). There was one case of posttransplant lymphoproliferative disease (PTLD), and no data on LCH recurrences. The authors conducted a literature review to identify an additional 50 patients with LCH who underwent a liver transplant. Of these patients, 47% experienced rejection, 11% had PTLD, and 8% had recurrent LCH. Seven patients (14%) with graft loss were treated with retransplant.[159][Level of evidence C2]
Case reports and case series have documented the efficacy of MAPK inhibitors for the treatment of progressive hepatic LCH.[148,160]
Some patients develop a macrophage activation of their marrow. This could be confusing to clinicians, who may think the patient has hemophagocytic lymphohistiocytosis (HLH) and LCH. The best therapy for this life-threatening manifestation is not clear because it tends not to respond well to standard HLH therapy. Clofarabine, anti-CD52 antibody alemtuzumab, or reduced-intensity allogeneic stem cell transplant could be considered.[161][Level of evidence C3] It is unknown whether newer HLH therapies, such as the antibody to interferon-gamma or the JAK-STAT inhibitor ruxolitinib, will be more effective in the LCH-macrophage activation than the above options.
Treatment options under clinical evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- NCT04079179 (Cobimetinib for the Treatment of Refractory LCH): This study is open to children or adults with relapsed or refractory LCH or other newly diagnosed, relapsed, or refractory histiocytic disorders.
Assessment of Response to Treatment
Response assessment remains one of the most difficult areas in LCH therapy. It is easier when there is a specific area that can be monitored clinically or with ultrasonography, CT, PET, or MRI scans, such as the skin, hepato/splenomegaly, and other mass or lytic bone lesions. Clinical judgment, including evaluation of pain and other symptoms, remains important.
Bone lesions may take many months to heal and are difficult to evaluate on plain radiographs, although sclerosis around the periphery of a bone lesion suggests healing. CT or MRI scans are useful in assessing response of a soft tissue mass associated with a bone lesion, but are not particularly helpful in assessing the response of lytic bone lesions. Technetium Tc 99m bone scans remain positive in healing bone. PET scans may be helpful in monitoring the response to therapy because the intensity of the PET image diminishes with the response of lesions and healing of bone.[17]
For children or adults with lung LCH, pulmonary function testing and high-resolution CT scans are sensitive methods for detecting disease progression.[20] Residual interstitial changes reflecting residual fibrosis or residual inactive cysts must be distinguished from active disease; somatostatin analog scintigraphy may be useful in this regard.[162]
Treatment Options No Longer Considered Effective for Childhood LCH
Treatments that have been used in the past but are no longer recommended for pediatric patients with LCH include cyclosporine [163] and interferon-alpha.[164]
Extensive surgery is also not indicated. For lesions of the mandible, extensive surgery may destroy any possibility of secondary tooth development. Surgical resection of groin or genital lesions is contraindicated because these lesions can be healed by chemotherapy.
Radiation therapy use in LCH has been significantly reduced in pediatric patients, and even low-dose radiation therapy should be limited to single-bone, vertebral body lesions or other single-bone lesions compressing the spinal cord or optic nerve that do not respond to chemotherapy or are painful and not amenable to other therapy.[94,101,165]
Late Disease and Treatment Effects of Childhood LCH
The reported frequency of long-term consequences of LCH has ranged from 20% to 70%. Children with low–risk-organ involvement (skin, bones, lymph nodes, or pituitary gland) have an approximately 20% chance of developing long-term sequelae.[30,166]; [167][Level of evidence B4] Patients with multisystem involvement have a reported rate of long-term complications of approximately 70% when treatment was only 6 months.[30,101,168,169] However, the extent of long-term sequelae in patients who are treated for a year has not been reported.
This wide variation in frequency results from case definition, sample size, therapy used, method of data collection, and follow-up duration. Quality-of-life studies have reported the following:
- In one study of long-term survivors of skeletal LCH, the quality-of-life scores were not significantly different from those of healthy control children and adults.[104] In addition, the quality-of-life scores were very similar between those with and without permanent sequelae.
- In another study of 40 patients who were carefully screened for late effects, adverse quality-of-life scores were found in more than 50% of patients.[40] Seventy-five percent of patients had detectable long-term sequelae. Hypothalamic/pituitary dysfunction (50%), cognitive dysfunction (20%), and cerebellar involvement (17.5%) were the most common side effects.
The late effects of LCH may occur in the following body systems:
- Endocrine. Patients with diabetes insipidus are at risk of panhypopituitarism and should be monitored carefully for adequacy of growth and development. In a retrospective review of 141 patients with LCH and diabetes insipidus, 43% developed growth hormone (GH) deficiency.[101,168,169] The 5-year risk of GH deficiency among children with LCH and diabetes insipidus was 35%, and the 10-year risk was 54%. There was no increased reactivation of LCH in patients who received GH compared with those who did not.[168] Growth and development problems are more frequent because of the young age at presentation and the more toxic effects of long-term prednisone therapy in the very young child.
- Special senses (hearing loss). Hearing loss has been found in 38% of children who were treated for LCH.[101] Seventy percent of patients with LCH in this study had ear involvement, which included aural discharge, mastoid swelling, and hearing loss. Of those with CT or MRI abnormalities in the mastoid, 59% had hearing loss.[170][Level of evidence C1]
- Neurological. Neurological symptoms secondary to vertebral compression of cervical lesions have been reported in 3 of 26 patients with LCH and spinal lesions.[101] CNS LCH occurs most often in children with LCH of the pituitary or CNS-risk skull bones (mastoid, orbit, or temporal bone). Significant cognitive defects and MRI abnormalities may develop in some long-term survivors with CNS-risk skull lesions.[171] Some patients have markedly abnormal cerebellar function and behavior abnormalities, while others have subtle deficits in short-term memory and brain stem–evoked potentials.[83]
- Skeletal. Orthopedic problems from lesions of the spine, femur, tibia, or humerus may be seen in 20% of patients. These problems include vertebral collapse or instability of the spine that may lead to scoliosis and facial or limb asymmetry.
- Respiratory. Diffuse pulmonary disease may result in poor lung function with higher risk of infections and decreased exercise tolerance. These patients should be monitored with pulmonary function testing, including the diffusing capacity of carbon monoxide and ratio of residual volume to total lung capacity.[39]
- Digestive. Liver disease may lead to sclerosing cholangitis, which rarely responds to any treatment other than liver transplant.[129] Dental problems characterized by loss of teeth have been significant for some patients, usually related to overly aggressive dental surgery.[172]
-
Subsequent neoplasms. Bone marrow failure secondary to LCH or from therapy is rare and is associated with a higher risk of malignancy. Patients with LCH have a higher-than-normal risk of developing secondary cancers.[173,174]
Leukemia (usually acute myeloid leukemia) occurs after treatment, as does lymphoblastic lymphoma. Concurrent LCH and malignancy has been reported in a few patients, and some patients had their malignancy first, followed by development of LCH. Three patients with T-cell acute lymphoblastic leukemia (ALL) and aggressive LCH were reported and, as with all histiocytic disorders associated with or following lymphoblastic malignancies, the same genetic changes were found in both diseases, suggesting a shared clonal origin.[175,176,177] One study reported two cases in which clonality with the same T-cell receptor gamma genotype was found.[176] The authors of this study emphasized the plasticity of lymphocytes developing into Langerhans cells. The second study described one patient with LCH after T-cell ALL who had the same T-cell receptor gene rearrangements and activating variants of the NOTCH1 gene.[177]
A publication based on surveying Histiocyte Society members and a literature review reported 116 cases of childhood LCH-malignancy pairs. Leukemias and myeloproliferative disorders (n = 58; 50.0%) prevailed over solid tumors (n = 43; 37.1%) and lymphomas (n = 15; 12.9%). In most children, malignancy followed LCH (n = 69; 59.5%). However, ALL, including T-cell ALL, was sometimes seen preceding the onset of LCH or histiocytic neoplasms. The histiocytic disorder commonly carried the same underlying genetic findings as the preceding leukemia.[178]
Another study reported a population-based analysis of subsequent malignancies in pediatric patients in the Surveillance, Epidemiology, and End Results (SEER) Program database from 2000 to 2016.[179] Of the 936 pediatric cases, there were 2 cases of non-Hodgkin lymphoma, 2 cases of Hodgkin lymphoma, and 1 case of T-cell ALL. However, the median follow-up was 38 months, which may not be sufficient to capture secondary solid tumors.
References:
- Carstensen H, Ornvold K: The epidemiology of Langerhans cell histiocytosis in children in Denmark, 1975-89. [Abstract] Med Pediatr Oncol 21 (5): A-15, 387-8, 1993.
- Salotti JA, Nanduri V, Pearce MS, et al.: Incidence and clinical features of Langerhans cell histiocytosis in the UK and Ireland. Arch Dis Child 94 (5): 376-80, 2009.
- Stålemark H, Laurencikas E, Karis J, et al.: Incidence of Langerhans cell histiocytosis in children: a population-based study. Pediatr Blood Cancer 51 (1): 76-81, 2008.
- A multicentre retrospective survey of Langerhans' cell histiocytosis: 348 cases observed between 1983 and 1993. The French Langerhans' Cell Histiocytosis Study Group. Arch Dis Child 75 (1): 17-24, 1996.
- Guyot-Goubin A, Donadieu J, Barkaoui M, et al.: Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000-2004. Pediatr Blood Cancer 51 (1): 71-5, 2008.
- Liu H, Stiller CA, Crooks CJ, et al.: Incidence, prevalence and survival in patients with Langerhans cell histiocytosis: A national registry study from England, 2013-2019. Br J Haematol 199 (5): 728-738, 2022.
- Ribeiro KB, Degar B, Antoneli CB, et al.: Ethnicity, race, and socioeconomic status influence incidence of Langerhans cell histiocytosis. Pediatr Blood Cancer 62 (6): 982-7, 2015.
- Bhatia S, Nesbit ME, Egeler RM, et al.: Epidemiologic study of Langerhans cell histiocytosis in children. J Pediatr 130 (5): 774-84, 1997.
- Peckham-Gregory EC, Danysh HE, Brown AL, et al.: Evaluation of maternal and perinatal characteristics on childhood lymphoma risk: A population-based case-control study. Pediatr Blood Cancer 64 (5): , 2017.
- Peckham-Gregory EC, Chakraborty R, Scheurer ME, et al.: A genome-wide association study of LCH identifies a variant in SMAD6 associated with susceptibility. Blood 130 (20): 2229-2232, 2017.
- Venkatramani R, Rosenberg S, Indramohan G, et al.: An exploratory epidemiological study of Langerhans cell histiocytosis. Pediatr Blood Cancer 59 (7): 1324-6, 2012.
- Nicholson HS, Egeler RM, Nesbit ME: The epidemiology of Langerhans cell histiocytosis. Hematol Oncol Clin North Am 12 (2): 379-84, 1998.
- McClain K, Jin H, Gresik V, et al.: Langerhans cell histiocytosis: lack of a viral etiology. Am J Hematol 47 (1): 16-20, 1994.
- Jeziorski E, Senechal B, Molina TJ, et al.: Herpes-virus infection in patients with Langerhans cell histiocytosis: a case-controlled sero-epidemiological study, and in situ analysis. PLoS One 3 (9): e3262, 2008.
- Haupt R, Minkov M, Astigarraga I, et al.: Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer 60 (2): 175-84, 2013.
- Binkovitz LA, Olshefski RS, Adler BH: Coincidence FDG-PET in the evaluation of Langerhans' cell histiocytosis: preliminary findings. Pediatr Radiol 33 (9): 598-602, 2003.
- Phillips M, Allen C, Gerson P, et al.: Comparison of FDG-PET scans to conventional radiography and bone scans in management of Langerhans cell histiocytosis. Pediatr Blood Cancer 52 (1): 97-101, 2009.
- Ribeiro MJ, Idbaih A, Thomas C, et al.: 18F-FDG PET in neurodegenerative Langerhans cell histiocytosis : results and potential interest for an early diagnosis of the disease. J Neurol 255 (4): 575-80, 2008.
- Grois N, Prayer D, Prosch H, et al.: Course and clinical impact of magnetic resonance imaging findings in diabetes insipidus associated with Langerhans cell histiocytosis. Pediatr Blood Cancer 43 (1): 59-65, 2004.
- Ha SY, Helms P, Fletcher M, et al.: Lung involvement in Langerhans' cell histiocytosis: prevalence, clinical features, and outcome. Pediatrics 89 (3): 466-9, 1992.
- Prasad SR, Wang H, Rosas H, et al.: Fat-containing lesions of the liver: radiologic-pathologic correlation. Radiographics 25 (2): 321-31, 2005 Mar-Apr.
- Ferrell J, Sharp S, Kumar A, et al.: Discrepancies between F-18-FDG PET/CT findings and conventional imaging in Langerhans cell histiocytosis. Pediatr Blood Cancer 68 (4): e28891, 2021.
- Rameh V, Voss S, Bedoya MA, et al.: The added value of skeletal surveys in the initial evaluation of children diagnosed with Langerhans cell histiocytosis in the era of staging 18 F-FDG PET/CT: A retrospective study. Pediatr Blood Cancer 70 (1): e30057, 2023.
- An R, Ma X, Wang Y: The value of 18F-FDG PET/CT in Langerhans cell histiocytosis. Ann Nucl Med 38 (3): 238-245, 2024.
- Prayer D, Grois N, Prosch H, et al.: MR imaging presentation of intracranial disease associated with Langerhans cell histiocytosis. AJNR Am J Neuroradiol 25 (5): 880-91, 2004.
- Ronceray L, Pötschger U, Janka G, et al.: Pulmonary involvement in pediatric-onset multisystem Langerhans cell histiocytosis: effect on course and outcome. J Pediatr 161 (1): 129-33.e1-3, 2012.
- Gadner H, Grois N, Arico M, et al.: A randomized trial of treatment for multisystem Langerhans' cell histiocytosis. J Pediatr 138 (5): 728-34, 2001.
- Gadner H, Grois N, Pötschger U, et al.: Improved outcome in multisystem Langerhans cell histiocytosis is associated with therapy intensification. Blood 111 (5): 2556-62, 2008.
- Gadner H, Minkov M, Grois N, et al.: Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis. Blood 121 (25): 5006-14, 2013.
- Haupt R, Nanduri V, Calevo MG, et al.: Permanent consequences in Langerhans cell histiocytosis patients: a pilot study from the Histiocyte Society-Late Effects Study Group. Pediatr Blood Cancer 42 (5): 438-44, 2004.
- Minkov M, Prosch H, Steiner M, et al.: Langerhans cell histiocytosis in neonates. Pediatr Blood Cancer 45 (6): 802-7, 2005.
- Héritier S, Emile JF, Barkaoui MA, et al.: BRAF Mutation Correlates With High-Risk Langerhans Cell Histiocytosis and Increased Resistance to First-Line Therapy. J Clin Oncol 34 (25): 3023-30, 2016.
- Berres ML, Lim KP, Peters T, et al.: BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med 211 (4): 669-83, 2014.
- Kemps PG, Zondag TCE, Arnardóttir HB, et al.: Clinicogenomic associations in childhood Langerhans cell histiocytosis: an international cohort study. Blood Adv 7 (4): 664-679, 2023.
- Rodriguez-Galindo C, Allen CE: Langerhans cell histiocytosis. Blood 135 (16): 1319-1331, 2020.
- Wnorowski M, Prosch H, Prayer D, et al.: Pattern and course of neurodegeneration in Langerhans cell histiocytosis. J Pediatr 153 (1): 127-32, 2008.
- Yeh EA, Greenberg J, Abla O, et al.: Evaluation and treatment of Langerhans cell histiocytosis patients with central nervous system abnormalities: Current views and new vistas. Pediatr Blood Cancer 65 (1): , 2018.
- Allen CE, Flores R, Rauch R, et al.: Neurodegenerative central nervous system Langerhans cell histiocytosis and coincident hydrocephalus treated with vincristine/cytosine arabinoside. Pediatr Blood Cancer 54 (3): 416-23, 2010.
- Bernstrand C, Cederlund K, Henter JI: Pulmonary function testing and pulmonary Langerhans cell histiocytosis. Pediatr Blood Cancer 49 (3): 323-8, 2007.
- Nanduri VR, Pritchard J, Levitt G, et al.: Long term morbidity and health related quality of life after multi-system Langerhans cell histiocytosis. Eur J Cancer 42 (15): 2563-9, 2006.
- Smith MA, Seibel NL, Altekruse SF, et al.: Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 28 (15): 2625-34, 2010.
- American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed February 25, 2025.
- Minkov M, Pötschger U, Thacker N, et al.: Additive Prognostic Impact of Gastrointestinal Involvement in Severe Multisystem Langerhans Cell Histiocytosis. J Pediatr 237: 65-70.e3, 2021.
- Donadieu J, Egeler RM, Pritchard J: Langerhans cell histiocytosis: a clinical update. In: Weitzman S, Egeler R M, eds.: Histiocytic Disorders of Children and Adults. Cambridge, United Kingdom: Cambridge University Press, 2005, pp 95-129.
- Slater JM, Swarm OJ: Eosinophilic granuloma of bone. Med Pediatr Oncol 8 (2): 151-64, 1980.
- Peng XS, Pan T, Chen LY, et al.: Langerhans' cell histiocytosis of the spine in children with soft tissue extension and chemotherapy. Int Orthop 33 (3): 731-6, 2009.
- Boztug K, Frimpong-Ansah K, Nanduri VR, et al.: Intraocular Langerhans cell histiocytosis in a neonate resulting in bilateral loss of vision. Pediatr Blood Cancer 47 (5): 633-5, 2006.
- Simko SJ, Garmezy B, Abhyankar H, et al.: Differentiating skin-limited and multisystem Langerhans cell histiocytosis. J Pediatr 165 (5): 990-6, 2014.
- Stein SL, Paller AS, Haut PR, et al.: Langerhans cell histiocytosis presenting in the neonatal period: a retrospective case series. Arch Pediatr Adolesc Med 155 (7): 778-83, 2001.
- Lau L, Krafchik B, Trebo MM, et al.: Cutaneous Langerhans cell histiocytosis in children under one year. Pediatr Blood Cancer 46 (1): 66-71, 2006.
- Munn S, Chu AC: Langerhans cell histiocytosis of the skin. Hematol Oncol Clin North Am 12 (2): 269-86, 1998.
- Hashimoto K, Griffin D, Kohsbaki M: Self-healing reticulohistiocytosis: a clinical, histologic, and ultrastructural study of the fourth case in the literature. Cancer 49 (2): 331-7, 1982.
- Ashena Z, Alavi S, Arzanian MT, et al.: Nail involvement in langerhans cell histiocytosis. Pediatr Hematol Oncol 24 (1): 45-51, 2007 Jan-Feb.
- Madrigal-Martínez-Pereda C, Guerrero-Rodríguez V, Guisado-Moya B, et al.: Langerhans cell histiocytosis: literature review and descriptive analysis of oral manifestations. Med Oral Patol Oral Cir Bucal 14 (5): E222-8, 2009.
- Hicks J, Flaitz CM: Langerhans cell histiocytosis: current insights in a molecular age with emphasis on clinical oral and maxillofacial pathology practice. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 100 (2 Suppl): S42-66, 2005.
- Ducassou S, Seyrig F, Thomas C, et al.: Thymus and mediastinal node involvement in childhood Langerhans cell histiocytosis: long-term follow-up from the French national cohort. Pediatr Blood Cancer 60 (11): 1759-65, 2013.
- Vassallo R, Ryu JH, Colby TV, et al.: Pulmonary Langerhans'-cell histiocytosis. N Engl J Med 342 (26): 1969-78, 2000.
- Le Louet S, Barkaoui MA, Miron J, et al.: Childhood Langerhans cell histiocytosis with severe lung involvement: a nationwide cohort study. Orphanet J Rare Dis 15 (1): 241, 2020.
- Odame I, Li P, Lau L, et al.: Pulmonary Langerhans cell histiocytosis: a variable disease in childhood. Pediatr Blood Cancer 47 (7): 889-93, 2006.
- Abbritti M, Mazzei MA, Bargagli E, et al.: Utility of spiral CAT scan in the follow-up of patients with pulmonary Langerhans cell histiocytosis. Eur J Radiol 81 (8): 1907-12, 2012.
- Burnett A, Carney D, Mukhopadhyay S, et al.: Thyroid involvement with Langerhans cell histiocytosis in a 3-year-old male. Pediatr Blood Cancer 50 (3): 726-7, 2008.
- Imashuku S, Kinugawa N, Matsuzaki A, et al.: Langerhans cell histiocytosis with multifocal bone lesions: comparative clinical features between single and multi-systems. Int J Hematol 90 (4): 506-12, 2009.
- Aricò M, Astigarraga I, Braier J, et al.: Lack of bone lesions at diagnosis is associated with inferior outcome in multisystem langerhans cell histiocytosis of childhood. Br J Haematol 169 (2): 241-8, 2015.
- Goyal R, Das A, Nijhawan R, et al.: Langerhans cell histiocytosis infiltration into pancreas and kidney. Pediatr Blood Cancer 49 (5): 748-50, 2007.
- Hait E, Liang M, Degar B, et al.: Gastrointestinal tract involvement in Langerhans cell histiocytosis: case report and literature review. Pediatrics 118 (5): e1593-9, 2006.
- Geissmann F, Thomas C, Emile JF, et al.: Digestive tract involvement in Langerhans cell histiocytosis. The French Langerhans Cell Histiocytosis Study Group. J Pediatr 129 (6): 836-45, 1996.
- Donadieu J, Rolon MA, Thomas C, et al.: Endocrine involvement in pediatric-onset Langerhans' cell histiocytosis: a population-based study. J Pediatr 144 (3): 344-50, 2004.
- Robison NJ, Prabhu SP, Sun P, et al.: Predictors of neoplastic disease in children with isolated pituitary stalk thickening. Pediatr Blood Cancer 60 (10): 1630-5, 2013.
- Prosch H, Grois N, Prayer D, et al.: Central diabetes insipidus as presenting symptom of Langerhans cell histiocytosis. Pediatr Blood Cancer 43 (5): 594-9, 2004.
- Richards GE, Thomsett MJ, Boston BA, et al.: Natural history of idiopathic diabetes insipidus. J Pediatr 159 (4): 566-70, 2011.
- Di Iorgi N, Allegri AE, Napoli F, et al.: Central diabetes insipidus in children and young adults: etiological diagnosis and long-term outcome of idiopathic cases. J Clin Endocrinol Metab 99 (4): 1264-72, 2014.
- Marchand I, Barkaoui MA, Garel C, et al.: Central diabetes insipidus as the inaugural manifestation of Langerhans cell histiocytosis: natural history and medical evaluation of 26 children and adolescents. J Clin Endocrinol Metab 96 (9): E1352-60, 2011.
- Grois N, Pötschger U, Prosch H, et al.: Risk factors for diabetes insipidus in langerhans cell histiocytosis. Pediatr Blood Cancer 46 (2): 228-33, 2006.
- Shioda Y, Adachi S, Imashuku S, et al.: Analysis of 43 cases of Langerhans cell histiocytosis (LCH)-induced central diabetes insipidus registered in the JLSG-96 and JLSG-02 studies in Japan. Int J Hematol 94 (6): 545-51, 2011.
- Sakamoto K, Morimoto A, Shioda Y, et al.: Central diabetes insipidus in pediatric patients with Langerhans cell histiocytosis: Results from the JLSG-96/02 studies. Pediatr Blood Cancer 66 (1): e27454, 2019.
- Gadner H, Heitger A, Grois N, et al.: Treatment strategy for disseminated Langerhans cell histiocytosis. DAL HX-83 Study Group. Med Pediatr Oncol 23 (2): 72-80, 1994.
- Dunger DB, Broadbent V, Yeoman E, et al.: The frequency and natural history of diabetes insipidus in children with Langerhans-cell histiocytosis. N Engl J Med 321 (17): 1157-62, 1989.
- Grois NG, Favara BE, Mostbeck GH, et al.: Central nervous system disease in Langerhans cell histiocytosis. Hematol Oncol Clin North Am 12 (2): 287-305, 1998.
- Grois N, Prayer D, Prosch H, et al.: Neuropathology of CNS disease in Langerhans cell histiocytosis. Brain 128 (Pt 4): 829-38, 2005.
- Fahrner B, Prosch H, Minkov M, et al.: Long-term outcome of hypothalamic pituitary tumors in Langerhans cell histiocytosis. Pediatr Blood Cancer 58 (4): 606-10, 2012.
- Héritier S, Barkaoui MA, Miron J, et al.: Incidence and risk factors for clinical neurodegenerative Langerhans cell histiocytosis: a longitudinal cohort study. Br J Haematol 183 (4): 608-617, 2018.
- McClain KL, Picarsic J, Chakraborty R, et al.: CNS Langerhans cell histiocytosis: Common hematopoietic origin for LCH-associated neurodegeneration and mass lesions. Cancer 124 (12): 2607-2620, 2018.
- Mittheisz E, Seidl R, Prayer D, et al.: Central nervous system-related permanent consequences in patients with Langerhans cell histiocytosis. Pediatr Blood Cancer 48 (1): 50-6, 2007.
- Minkov M, Steiner M, Pötschger U, et al.: Reactivations in multisystem Langerhans cell histiocytosis: data of the international LCH registry. J Pediatr 153 (5): 700-5, 705.e1-2, 2008.
- Braier JL, Rosso D, Latella A, et al.: Importance of multi-lineage hematologic involvement and hypoalbuminemia at diagnosis in patients with "risk-organ" multi-system Langerhans cell histiocytosis. J Pediatr Hematol Oncol 32 (4): e122-5, 2010.
- Steen AE, Steen KH, Bauer R, et al.: Successful treatment of cutaneous Langerhans cell histiocytosis with low-dose methotrexate. Br J Dermatol 145 (1): 137-40, 2001.
- Zinn DJ, Grimes AB, Lin H, et al.: Hydroxyurea: a new old therapy for Langerhans cell histiocytosis. Blood 128 (20): 2462-2465, 2016.
- McClain KL, Kozinetz CA: A phase II trial using thalidomide for Langerhans cell histiocytosis. Pediatr Blood Cancer 48 (1): 44-9, 2007.
- Uppuluri R, Ramachandrakurup S, Subburaj D, et al.: Excellent remission rates with limited toxicity in relapsed/refractory Langerhans cell histiocytosis with pulse dexamethasone and lenalidomide in children. Pediatr Blood Cancer 64 (1): 110-112, 2017.
- Hoeger PH, Nanduri VR, Harper JI, et al.: Long term follow up of topical mustine treatment for cutaneous langerhans cell histiocytosis. Arch Dis Child 82 (6): 483-7, 2000.
- Lindahl LM, Fenger-Grøn M, Iversen L: Topical nitrogen mustard therapy in patients with Langerhans cell histiocytosis. Br J Dermatol 166 (3): 642-5, 2012.
- Kwon OS, Cho KH, Song KY: Primary cutaneous Langerhans cell histiocytosis treated with photochemotherapy. J Dermatol 24 (1): 54-6, 1997.
- Vogel CA, Aughenbaugh W, Sharata H: Excimer laser as adjuvant therapy for adult cutaneous Langerhans cell histiocytosis. Arch Dermatol 144 (10): 1287-90, 2008.
- Laird J, Ma J, Chau K, et al.: Outcome After Radiation Therapy for Langerhans Cell Histiocytosis Is Dependent on Site of Involvement. Int J Radiat Oncol Biol Phys 100 (3): 670-678, 2018.
- Selch MT, Parker RG: Radiation therapy in the management of Langerhans cell histiocytosis. Med Pediatr Oncol 18 (2): 97-102, 1990.
- Kotecha R, Venkatramani R, Jubran RF, et al.: Clinical outcomes of radiation therapy in the management of Langerhans cell histiocytosis. Am J Clin Oncol 37 (6): 592-6, 2014.
- Greenberger JS, Cassady JR, Jaffe N, et al.: Radiation therapy in patients with histiocytosis: management of diabetes insipidus and bone lesions. Int J Radiat Oncol Biol Phys 5 (10): 1749-55, 1979.
- Nauert C, Zornoza J, Ayala A, et al.: Eosinophilic granuloma of bone: diagnosis and management. Skeletal Radiol 10 (4): 227-35, 1983.
- Gramatovici R, D'Angio GJ: Radiation therapy in soft-tissue lesions in histiocytosis X (Langerhans' cell histiocytosis). Med Pediatr Oncol 16 (4): 259-62, 1988.
- Baptista AM, Camargo AF, de Camargo OP, et al.: Does adjunctive chemotherapy reduce remission rates compared to cortisone alone in unifocal or multifocal histiocytosis of bone? Clin Orthop Relat Res 470 (3): 663-9, 2012.
- Willis B, Ablin A, Weinberg V, et al.: Disease course and late sequelae of Langerhans' cell histiocytosis: 25-year experience at the University of California, San Francisco. J Clin Oncol 14 (7): 2073-82, 1996.
- Woo KI, Harris GJ: Eosinophilic granuloma of the orbit: understanding the paradox of aggressive destruction responsive to minimal intervention. Ophthal Plast Reconstr Surg 19 (6): 429-39, 2003.
- Gatineau-Sailliant S, Grimard P, Miron MC, et al.: Langerhans Cell Histiocytosis With Vertebral Involvement Diagnosed and Treated Over the Last 15 Years in a Single Canadian Pediatric Academic Institution. J Pediatr Hematol Oncol 42 (3): 222-227, 2020.
- Lau LM, Stuurman K, Weitzman S: Skeletal Langerhans cell histiocytosis in children: permanent consequences and health-related quality of life in long-term survivors. Pediatr Blood Cancer 50 (3): 607-12, 2008.
- Mammano S, Candiotto S, Balsano M: Cast and brace treatment of eosinophilic granuloma of the spine: long-term follow-up. J Pediatr Orthop 17 (6): 821-7, 1997 Nov-Dec.
- Titgemeyer C, Grois N, Minkov M, et al.: Pattern and course of single-system disease in Langerhans cell histiocytosis data from the DAL-HX 83- and 90-study. Med Pediatr Oncol 37 (2): 108-14, 2001.
- Morimoto A, Shioda Y, Imamura T, et al.: Intensification of induction therapy and prolongation of maintenance therapy did not improve the outcome of pediatric Langerhans cell histiocytosis with single-system multifocal bone lesions: results of the Japan Langerhans Cell Histiocytosis Study Group-02 Protocol Study. Int J Hematol 108 (2): 192-198, 2018.
- Egeler RM, de Kraker J, Voûte PA: Cytosine-arabinoside, vincristine, and prednisolone in the treatment of children with disseminated Langerhans cell histiocytosis with organ dysfunction: experience at a single institution. Med Pediatr Oncol 21 (4): 265-70, 1993.
- Weitzman S, Braier J, Donadieu J, et al.: 2'-Chlorodeoxyadenosine (2-CdA) as salvage therapy for Langerhans cell histiocytosis (LCH). results of the LCH-S-98 protocol of the Histiocyte Society. Pediatr Blood Cancer 53 (7): 1271-6, 2009.
- Farran RP, Zaretski E, Egeler RM: Treatment of Langerhans cell histiocytosis with pamidronate. J Pediatr Hematol Oncol 23 (1): 54-6, 2001.
- Chellapandian D, Makras P, Kaltsas G, et al.: Bisphosphonates in Langerhans Cell Histiocytosis: An International Retrospective Case Series. Mediterr J Hematol Infect Dis 8 (1): e2016033, 2016.
- Morimoto A, Shioda Y, Imamura T, et al.: Nationwide survey of bisphosphonate therapy for children with reactivated Langerhans cell histiocytosis in Japan. Pediatr Blood Cancer 56 (1): 110-5, 2011.
- Sivendran S, Harvey H, Lipton A, et al.: Treatment of Langerhans cell histiocytosis bone lesions with zoledronic acid: a case series. Int J Hematol 93 (6): 782-6, 2011.
- Büchler T, Cervinek L, Belohlavek O, et al.: Langerhans cell histiocytosis with central nervous system involvement: follow-up by FDG-PET during treatment with cladribine. Pediatr Blood Cancer 44 (3): 286-8, 2005.
- Watts J, Files B: Langerhans cell histiocytosis: central nervous system involvement treated successfully with 2-chlorodeoxyadenosine. Pediatr Hematol Oncol 18 (3): 199-204, 2001 Apr-May.
- Dhall G, Finlay JL, Dunkel IJ, et al.: Analysis of outcome for patients with mass lesions of the central nervous system due to Langerhans cell histiocytosis treated with 2-chlorodeoxyadenosine. Pediatr Blood Cancer 50 (1): 72-9, 2008.
- Grois N, Fahrner B, Arceci RJ, et al.: Central nervous system disease in Langerhans cell histiocytosis. J Pediatr 156 (6): 873-81, 881.e1, 2010.
- Ng Wing Tin S, Martin-Duverneuil N, Idbaih A, et al.: Efficacy of vinblastine in central nervous system Langerhans cell histiocytosis: a nationwide retrospective study. Orphanet J Rare Dis 6 (1): 83, 2011.
- Imashuku S, Arceci RJ: Strategies for the Prevention of Central Nervous System Complications in Patients with Langerhans Cell Histiocytosis: The Problem of Neurodegenerative Syndrome. Hematol Oncol Clin North Am 29 (5): 875-93, 2015.
- Idbaih A, Donadieu J, Barthez MA, et al.: Retinoic acid therapy in "degenerative-like" neuro-langerhans cell histiocytosis: a prospective pilot study. Pediatr Blood Cancer 43 (1): 55-8, 2004.
- Imashuku S, Ishida S, Koike K, et al.: Cerebellar ataxia in pediatric patients with Langerhans cell histiocytosis. J Pediatr Hematol Oncol 26 (11): 735-9, 2004.
- Imashuku S, Okazaki NA, Nakayama M, et al.: Treatment of neurodegenerative CNS disease in Langerhans cell histiocytosis with a combination of intravenous immunoglobulin and chemotherapy. Pediatr Blood Cancer 50 (2): 308-11, 2008.
- Chohan G, Barnett Y, Gibson J, et al.: Langerhans cell histiocytosis with refractory central nervous system involvement responsive to infliximab. J Neurol Neurosurg Psychiatry 83 (5): 573-5, 2012.
- Eckstein OS, Visser J, Rodriguez-Galindo C, et al.: Clinical responses and persistent BRAF V600E+ blood cells in children with LCH treated with MAPK pathway inhibition. Blood 133 (15): 1691-1694, 2019.
- Morimoto A, Ikushima S, Kinugawa N, et al.: Improved outcome in the treatment of pediatric multifocal Langerhans cell histiocytosis: Results from the Japan Langerhans Cell Histiocytosis Study Group-96 protocol study. Cancer 107 (3): 613-9, 2006.
- Eckstein O, McAtee CL, Greenberg J, et al.: Rituximab therapy for patients with Langerhans cell histiocytosis-associated neurologic dysfunction. Pediatr Hematol Oncol 35 (7-8): 427-433, 2018 Oct - Nov.
- Wong A, Ortiz-Neira CL, Reslan WA, et al.: Liver involvement in Langerhans cell histiocytosis. Pediatr Radiol 36 (10): 1105-7, 2006.
- Jaffe R: Liver involvement in the histiocytic disorders of childhood. Pediatr Dev Pathol 7 (3): 214-25, 2004 May-Jun.
- Braier J, Ciocca M, Latella A, et al.: Cholestasis, sclerosing cholangitis, and liver transplantation in Langerhans cell Histiocytosis. Med Pediatr Oncol 38 (3): 178-82, 2002.
- Carrere X, Pinto N, Gene Olaciregui N, et al.: High prevalence of BRAFV600E in patients with cholestasis, sclerosing cholangitis or liver fibrosis secondary to Langerhans cell histiocytosis. Pediatr Blood Cancer 68 (7): e29115, 2021.
- McClain K, Ramsay NK, Robison L, et al.: Bone marrow involvement in histiocytosis X. Med Pediatr Oncol 11 (3): 167-71, 1983.
- Minkov M, Pötschger U, Grois N, et al.: Bone marrow assessment in Langerhans cell histiocytosis. Pediatr Blood Cancer 49 (5): 694-8, 2007.
- Ballester LY, Cantu MD, Lim KPH, et al.: The use of BRAF V600E mutation-specific immunohistochemistry in pediatric Langerhans cell histiocytosis. Hematol Oncol 36 (1): 307-315, 2018.
- Galluzzo ML, Braier J, Rosenzweig SD, et al.: Bone marrow findings at diagnosis in patients with multisystem langerhans cell histiocytosis. Pediatr Dev Pathol 13 (2): 101-6, 2010 Mar-Apr.
- Favara BE, Jaffe R, Egeler RM: Macrophage activation and hemophagocytic syndrome in langerhans cell histiocytosis: report of 30 cases. Pediatr Dev Pathol 5 (2): 130-40, 2002 Mar-Apr.
- Morimoto A, Shioda Y, Imamura T, et al.: Intensified and prolonged therapy comprising cytarabine, vincristine and prednisolone improves outcome in patients with multisystem Langerhans cell histiocytosis: results of the Japan Langerhans Cell Histiocytosis Study Group-02 Protocol Study. Int J Hematol 104 (1): 99-109, 2016.
- Allen CE, Merad M, McClain KL: Langerhans-Cell Histiocytosis. N Engl J Med 379 (9): 856-868, 2018.
- Pollono D, Rey G, Latella A, et al.: Reactivation and risk of sequelae in Langerhans cell histiocytosis. Pediatr Blood Cancer 48 (7): 696-9, 2007.
- Simko SJ, McClain KL, Allen CE: Up-front therapy for LCH: is it time to test an alternative to vinblastine/prednisone? Br J Haematol 169 (2): 299-301, 2015.
- Barkaoui MA, Queheille E, Aladjidi N, et al.: Long-term follow-up of children with risk organ-negative Langerhans cell histiocytosis after 2-chlorodeoxyadenosine treatment. Br J Haematol 191 (5): 825-834, 2020.
- Simko SJ, Tran HD, Jones J, et al.: Clofarabine salvage therapy in refractory multifocal histiocytic disorders, including Langerhans cell histiocytosis, juvenile xanthogranuloma and Rosai-Dorfman disease. Pediatr Blood Cancer 61 (3): 479-87, 2014.
- Kudo K, Ohga S, Morimoto A, et al.: Improved outcome of refractory Langerhans cell histiocytosis in children with hematopoietic stem cell transplantation in Japan. Bone Marrow Transplant 45 (5): 901-6, 2010.
- Imamura T, Sato T, Shiota Y, et al.: Outcome of pediatric patients with Langerhans cell histiocytosis treated with 2 chlorodeoxyadenosine: a nationwide survey in Japan. Int J Hematol 91 (4): 646-51, 2010.
- Donadieu J, Bernard F, van Noesel M, et al.: Cladribine and cytarabine in refractory multisystem Langerhans cell histiocytosis: results of an international phase 2 study. Blood 126 (12): 1415-23, 2015.
- Rosso DA, Amaral D, Latella A, et al.: Reduced doses of cladribine and cytarabine regimen was effective and well tolerated in patients with refractory-risk multisystem Langerhans cell histiocytosis. Br J Haematol 172 (2): 287-90, 2016.
- Rodriguez-Galindo C, Jeng M, Khuu P, et al.: Clofarabine in refractory Langerhans cell histiocytosis. Pediatr Blood Cancer 51 (5): 703-6, 2008.
- Abraham A, Alsultan A, Jeng M, et al.: Clofarabine salvage therapy for refractory high-risk langerhans cell histiocytosis. Pediatr Blood Cancer 60 (6): E19-22, 2013.
- Donadieu J, Larabi IA, Tardieu M, et al.: Vemurafenib for Refractory Multisystem Langerhans Cell Histiocytosis in Children: An International Observational Study. J Clin Oncol 37 (31): 2857-2865, 2019.
- Tardieu M, Néron A, Duvert-Lehembre S, et al.: Cutaneous adverse events in children treated with vemurafenib for refractory BRAFV600E mutated Langerhans cell histiocytosis. Pediatr Blood Cancer 68 (9): e29140, 2021.
- Mohapatra D, Gupta AK, Haldar P, et al.: Efficacy and safety of vemurafenib in Langerhans cell histiocytosis (LCH): A systematic review and meta-analysis. Pediatr Hematol Oncol 40 (1): 86-97, 2023.
- Whitlock JA, Geoerger B, Dunkel IJ, et al.: Dabrafenib, alone or in combination with trametinib, in BRAF V600-mutated pediatric Langerhans cell histiocytosis. Blood Adv 7 (15): 3806-3815, 2023.
- Janku F, Amin HM, Yang D, et al.: Response of histiocytoses to imatinib mesylate: fire to ashes. J Clin Oncol 28 (31): e633-6, 2010.
- Wagner C, Mohme H, Krömer-Olbrisch T, et al.: Langerhans cell histiocytosis: treatment failure with imatinib. Arch Dermatol 145 (8): 949-50, 2009.
- Akkari V, Donadieu J, Piguet C, et al.: Hematopoietic stem cell transplantation in patients with severe Langerhans cell histiocytosis and hematological dysfunction: experience of the French Langerhans Cell Study Group. Bone Marrow Transplant 31 (12): 1097-103, 2003.
- Nagarajan R, Neglia J, Ramsay N, et al.: Successful treatment of refractory Langerhans cell histiocytosis with unrelated cord blood transplantation. J Pediatr Hematol Oncol 23 (9): 629-32, 2001.
- Caselli D, Aricò M; EBMT Paediatric Working Party: The role of BMT in childhood histiocytoses. Bone Marrow Transplant 41 (Suppl 2): S8-S13, 2008.
- Kudo K, Maeda M, Suzuki N, et al.: Nationwide retrospective review of hematopoietic stem cell transplantation in children with refractory Langerhans cell histiocytosis. Int J Hematol 111 (1): 137-148, 2020.
- Veys PA, Nanduri V, Baker KS, et al.: Haematopoietic stem cell transplantation for refractory Langerhans cell histiocytosis: outcome by intensity of conditioning. Br J Haematol 169 (5): 711-8, 2015.
- Ziogas IA, Kakos CD, Wu WK, et al.: Liver Transplantation for Langerhans Cell Histiocytosis: A US Population-Based Analysis and Systematic Review of the Literature. Liver Transpl 27 (8): 1181-1190, 2021.
- Lee LH, Krupski C, Clark J, et al.: High-risk LCH in infants is serially transplantable in a xenograft model but responds durably to targeted therapy. Blood Adv 4 (4): 717-727, 2020.
- Jordan MB, McClain KL, Yan X, et al.: Anti-CD52 antibody, alemtuzumab, binds to Langerhans cells in Langerhans cell histiocytosis. Pediatr Blood Cancer 44 (3): 251-4, 2005.
- Tazi A, Hiltermann J, Vassallo R: Adult lung histiocytosis. In: Weitzman S, Egeler R M, eds.: Histiocytic Disorders of Children and Adults. Cambridge, United Kingdom: Cambridge University Press, 2005, pp 187-207.
- Minkov M, Grois N, Broadbent V, et al.: Cyclosporine A therapy for multisystem langerhans cell histiocytosis. Med Pediatr Oncol 33 (5): 482-5, 1999.
- Lukina EA, Kuznetsov VP, Beliaev DL, et al.: [The treatment of histiocytosis X (Langerhans-cell histiocytosis) with alpha-interferon preparations] Ter Arkh 65 (11): 67-70, 1993.
- Gadner H, Ladisch S: The treatment of Langerhans cell histiocytosis. In: Weitzman S, Egeler R M, eds.: Histiocytic Disorders of Children and Adults. Cambridge, United Kingdom: Cambridge University Press, 2005, pp 229-53.
- Chow TW, Leung WK, Cheng FWT, et al.: Late outcomes in children with Langerhans cell histiocytosis. Arch Dis Child 102 (9): 830-835, 2017.
- Sakamoto K, Morimoto A, Shioda Y, et al.: Long-term complications in uniformly treated paediatric Langerhans histiocytosis patients disclosed by 12 years of follow-up of the JLSG-96/02 studies. Br J Haematol 192 (3): 615-620, 2021.
- Donadieu J, Rolon MA, Pion I, et al.: Incidence of growth hormone deficiency in pediatric-onset Langerhans cell histiocytosis: efficacy and safety of growth hormone treatment. J Clin Endocrinol Metab 89 (2): 604-9, 2004.
- Komp DM: Long-term sequelae of histiocytosis X. Am J Pediatr Hematol Oncol 3 (2): 163-8, 1981.
- Nanduri V, Tatevossian R, Sirimanna T: High incidence of hearing loss in long-term survivors of multisystem Langerhans cell histiocytosis. Pediatr Blood Cancer 54 (3): 449-53, 2010.
- Nanduri VR, Lillywhite L, Chapman C, et al.: Cognitive outcome of long-term survivors of multisystem langerhans cell histiocytosis: a single-institution, cross-sectional study. J Clin Oncol 21 (15): 2961-7, 2003.
- Guimarães LF, Dias PF, Janini ME, et al.: Langerhans cell histiocytosis: impact on the permanent dentition after an 8-year follow-up. J Dent Child (Chic) 75 (1): 64-8, 2008 Jan-Apr.
- Egeler RM, Neglia JP, Puccetti DM, et al.: Association of Langerhans cell histiocytosis with malignant neoplasms. Cancer 71 (3): 865-73, 1993.
- Egeler RM, Neglia JP, Aricò M, et al.: The relation of Langerhans cell histiocytosis to acute leukemia, lymphomas, and other solid tumors. The LCH-Malignancy Study Group of the Histiocyte Society. Hematol Oncol Clin North Am 12 (2): 369-78, 1998.
- Castro EC, Blazquez C, Boyd J, et al.: Clinicopathologic features of histiocytic lesions following ALL, with a review of the literature. Pediatr Dev Pathol 13 (3): 225-37, 2010 May-Jun.
- Feldman AL, Berthold F, Arceci RJ, et al.: Clonal relationship between precursor T-lymphoblastic leukaemia/lymphoma and Langerhans-cell histiocytosis. Lancet Oncol 6 (6): 435-7, 2005.
- Rodig SJ, Payne EG, Degar BA, et al.: Aggressive Langerhans cell histiocytosis following T-ALL: clonally related neoplasms with persistent expression of constitutively active NOTCH1. Am J Hematol 83 (2): 116-21, 2008.
- Bagnasco F, Zimmermann SY, Egeler RM, et al.: Langerhans cell histiocytosis and associated malignancies: A retrospective analysis of 270 patients. Eur J Cancer 172: 138-145, 2022.
- Goyal G, Parikh R, Richman J, et al.: Spectrum of second primary malignancies and cause-specific mortality in pediatric and adult langerhans cell histiocytosis. Leuk Res 126: 107032, 2023.
Adult LCH
The natural history of disease in adults with Langerhans cell histiocytosis (LCH) is poorly understood. Pulmonary LCH is the exception to this finding. Delays of many months or years commonly occur before adults are diagnosed, and they have long-term issues with chronic pain and fatigue. There are other differences from childhood LCH, including frequency of various bone sites of disease. It also appears that multisystem high-risk LCH in adults may be less aggressive than high-risk disease in children. A consensus group reported on the evaluation and treatment of adult patients with LCH.[1] However, treatment discussions continue, particularly regarding optimal first-line therapy.
A multicenter retrospective review of 219 adult patients (aged >18 years) with LCH was conducted to assess long-term outcomes. The median follow-up was 74 months. The 5-year disease-free survival rate was 58%, and the overall survival (OS) rate was 88%. About one-third of deaths were LCH-related and occurred within 5 years of diagnosis. Second cancers occurred in 16.4% of cases (both hematologic and solid tumors). Deaths that occurred 5 or more years after diagnosis were predominantly non-LCH related (i.e., second cancers, chronic obstructive pulmonary disease, and cardiovascular disease). Compared with the general U.S. population, patients with LCH had a higher standard mortality ratio (SMR) if diagnosed before age 55 years (SMR, 5.94) or had multisystem disease (SMR, 4.12).[2]
Incidence
A population-based study in England found that the incidence of LCH in patients older than 15 years was 1.05 cases per 1 million people.[3] Of these individuals, 44% were younger than 45 years. A higher incidence of LCH in economically disadvantaged areas was associated with a higher incidence of smoking in those areas.
More than 90% of adult pulmonary LCH cases occur in young adults who smoke, often more than 20 cigarettes per day.[4,5]
Clinical Presentation
Adult patients may have signs and symptoms of LCH for many months before receiving a definitive diagnosis and treatment. LCH in adults is often similar to that in children and appears to involve the same organs, although the incidence in each organ may be different. There is a predominance of lung disease in adults, usually occurring as single-system disease and closely associated with smoking and some unique biological characteristics. Most isolated lung LCH cases in adults are polyclonal and possibly reactive, while fewer lung LCH cases are monoclonal.[6,7]
A German registry with 121 registrants showed that 62% had single-organ involvement and 38% had multisystem involvement. Pulmonary LCH occurred in 34% of the total study population. Lungs are the most common site, followed by bone and skin involvement. The median age at diagnosis was 44 years (±12.8 years). All organ systems found in childhood LCH were seen in these adults, including endocrine and central nervous system (CNS), liver, spleen, bone marrow, and gastrointestinal tract. The major difference is the much higher incidence of isolated pulmonary LCH in adults, particularly in young adults who smoke. Other differences appear to be the more frequent involvement of genital and oral mucosa.[8]
Presenting signs and symptoms from published studies include the following:
- Dyspnea or tachypnea.
- Polydipsia and polyuria.
- Bone pain.
- Soft tissue swelling near bone lesions.
- Skin rash or scalp nodules.
- Lymphadenopathy.
- Weight loss.
- Fever.
- Gingival hypertrophy.
- Ataxia.
- Memory problems.
- Hepatosplenomegaly.
Patients who present with isolated diabetes insipidus should be carefully observed for the onset of other signs or symptoms characteristic of LCH. At least 80% of patients with diabetes insipidus had involvement of other organ systems, including bone (68%), skin (57%), lung (39%), and lymph nodes (18%).[9] However, isolated diabetes insipidus in adults is similar to that in pediatric patients, with progression from posterior to anterior pituitary/hypothalamus and to cerebellar involvement. For more information, see the Endocrine system section.
Skin and oral cavity
Thirty-seven percent of adults with multifocal LCH have skin involvement. Skin-only LCH occurs but it is less common in adults than in children. The prognosis for adults with skin-only LCH is excellent, with a 5-year survival probability of 100%. The cutaneous involvement is clinically similar to that seen in children and may take many forms.[10] Infra-mammary and vulvar involvement are frequent sites of presentation in adult women.
Many patients have a papular rash with brown, red, or crusted areas ranging from the size of a pinhead to a dime. In the scalp, the rash is similar to that of seborrhea. Skin in the inguinal region, genitalia, or around the anus may have open ulcers that do not heal after antibacterial or antifungal therapy. The lesions are usually asymptomatic but may be pruritic or painful. In the mouth, swollen gums or ulcers along the cheeks, soft or hard palate, gingiva, or tongue may be signs of LCH.
Diagnosis of LCH is usually made by skin biopsy performed for persistent skin lesions.[10]
Bone
The relative frequency of bone involvement in adults differs from that in children. The frequency of mandible involvement is 30% in adults and 7% in children, and the frequency of skull involvement is 21% in adults and 40% in children.[8,9,11,12] The frequencies of lesions in the vertebrae (13%), pelvis (13%), extremities (17%), and ribs (6%) in adults are similar to those found in children.[8]
Lung
Pulmonary LCH in adults (40%–50% of patients) is usually single-system disease. However, in some patients, other organs may be involved, including bone, skin, and hypothalamus/pituitary.[13]
Pulmonary LCH is more prevalent in smokers than in nonsmokers, and the male-to-female ratio is nearly 1:1, depending on the incidence of smoking in the population studied.[13,14] However, a study of pulmonary LCH from China reported that 73% of the patients were male.[15] Patients with pulmonary LCH usually present with a dry cough, dyspnea, or chest pain, although nearly 20% of adults with lung involvement have no symptoms.[16,17] Chest pain may indicate a spontaneous pneumothorax (10%–28% of adult pulmonary LCH cases).[15]
Pulmonary LCH can be diagnosed by bronchoscopy in about 50% of adult patients, as defined by immunostaining of at least 5% of CD1a-positive cells in the sample.[18] High-resolution lung computed tomography (CT) shows characteristic changes with cysts and nodules, more prevalent at the mid and upper zones. These findings have been characterized as pathognomonic for lung LCH.[16]
The LCH cells in adult lung lesions were shown to be mature dendritic cells expressing high levels of the accessory molecules CD80 and CD86, unlike Langerhans cells (LCs) found in other lung disorders.[17] MAPK pathway variants have been demonstrated in more than two-thirds of pulmonary LCH lesions in adults, suggesting a clonal process in a significant proportion of patients.[7,19]
In a review of 206 patients with pulmonary LCH from France (median follow-up, 5 years), the 10-year survival rate was 93%.[20] Patients who had chronic respiratory failure or pulmonary hypertension, both less than 5% of the study group, had much worse outcomes. Of these patients, 58% died. Patients with pulmonary LCH had a 17-fold higher incidence of lung carcinomas than an age- and sex-matched French population cohort.
Favorable prognostic factors for adult LCH of the lung include the following:
- Minimal symptoms. Adults with pulmonary LCH who have minimal symptoms have a good prognosis, although some have steady deterioration over many years.[5]
- Smoking cessation or treatment. Fifty-nine percent of patients do well with either spontaneous remission after cessation of smoking, or with some form of therapy.[5] However, one study reported that smoking cessation did not increase the longevity of adults with pulmonary LCH, apparently because the tempo of disease is so variable.[21] The authors of the review of 206 patients from France (see above) [20] noted that the two studies cited here had fewer patients, were retrospective, and did not perform high-resolution CT scans as frequently.[5,21] These older studies likely included patients with more severe disease than the French study.[20]
- Lung transplant. In one multicenter study, patients who received lung transplants for the treatment of pulmonary LCH had a 1-year survival rate of 77% and a 10-year survival rate of 54%, with a 20% chance of LCH recurrence.[22]
Unfavorable prognostic factors for adult LCH of the lung include the following:
- Altered pulmonary function. Lower forced expiratory volume/forced vital capacity (FEV1/FVC) ratio and higher residual volume/total lung capacity (RV/TLC) ratio are adverse prognostic variables.[21] Some patients have normal ventilatory function but abnormal carbon monoxide diffusion capacity.[15][Level of evidence C2] About 10% to 20% of patients have early severe progression to respiratory failure, severe pulmonary hypertension, and cor pulmonale. Adults who have progression with diffuse bullae formation, multiple pneumothoraces, and fibrosis have a poor prognosis.[23,24]; [15][Level of evidence C2]
- Age. Age older than 26 years is an adverse prognostic variable.[21]
- Smoking.
Most patients have a variable course, with stable disease in some patients and relapses and progression of respiratory dysfunction in others, often after many years.[25] A natural history study of 58 patients with pulmonary LCH found that 38% had deterioration of lung function after 2 years.[26] The most significant adverse prognostic variables were positive smoking statuses and low PaO2 levels at the time of inclusion.
The following results may be noted on diagnostic tests:
- Pulmonary function testing. The most frequent pulmonary function abnormality finding in patients with pulmonary LCH is a reduced carbon monoxide diffusing capacity, which is found in 70% to 90% of cases.[21,27]
- CT scan. A high-resolution CT scan, which reveals a reticulonodular pattern with cysts and nodules, usually in the upper lobes and sparing the costophrenic angle, is characteristic of LCH.[28] The presence of cystic abnormalities on high-resolution CT scans appears to be a poor predictor of which patients will have progressive disease.[29]
- Biopsy. Despite the typical CT findings, most pulmonologists agree that a lung biopsy is needed to confirm the diagnosis. A study that correlated lung CT findings and lung biopsy results in 27 patients with pulmonary LCH observed that thin-walled and bizarre cysts had active LCs and eosinophils.[30]
Liver
In one study, liver involvement was reported in 27% of adult patients with multiorgan disease.[31] Hepatomegaly (48%) and liver enzyme abnormalities (61%) were usually present. CT, magnetic resonance imaging (MRI), or ultrasonography imaging often find abnormalities along the biliary tract.
The early histopathological stage of liver LCH includes infiltration of CD1a-positive cells and periductal fibrosis with inflammatory infiltrates with or without steatosis. The late stage is biliary tree sclerosis. Treatment with ursodeoxycholic acid may be helpful.[31]
Endocrine system
Diabetes insipidus occurs in 25% of patients and may precede the diagnosis of LCH.[9] Anterior pituitary abnormalities are seen in approximately 20% of these patients.[32] Sometimes imaging studies of the pituitary are normal.[33]
Central nervous system (CNS)
The most frequent abnormalities in the CNS are enlargement of the pituitary, its stalk, and/or the hypothalamus. Brain involvement is typically in the cerebellum, pons, and basal ganglia, with abnormalities seen on the T2 and fluid-attenuated inversion recovery (FLAIR) images. Some patients have only imaging changes, but others have ataxia, dysmetria, dysarthria, and behavioral and psychological difficulties.[34]
Bone marrow and lymph nodes
Bone marrow involvement with LCH is uncommon and is usually heralded by abnormal blood counts, which could also be a sign of an underlying malignancy.[35] Lymph node infiltration in LCH is uncommon as an isolated finding, but can occur in up to 30% of patients with multisystem LCH.[34]
Gastrointestinal and cardiovascular systems
Gastrointestinal involvement is rare and usually presents with diarrhea and pain.[36] Abnormalities in the heart or around the great vessels often suggest a hybrid disease of Erdheim-Chester (ECD) and LCH.[37]
Multisystem disease
In a large series of patients from the Mayo Clinic, 31% had multisystem LCH, compared with 69% registered on the Histiocyte Society adult registry. This finding likely reflects referral bias.[10,38] In the adult patients with multisystem disease, the sites of disease included the following:
- Skin (50%).
- Mucocutaneous (40%).
- Pituitary/CNS (diabetes insipidus, 29.6%).
- Liver/spleen (hepatosplenomegaly, 16%).
- Thyroid (hypothyroidism, 6.6%).
- Lymph nodes (lymphadenopathy, 6%).
LCH and associated malignancies
Adult patients with LCH have higher rates of malignancies than do age-matched patients without LCH, by ratios of 2 to 4, depending on patient age.[39] A review of 132 patients with LCH from a single institution found 31 patients with other malignancies before their LCH diagnosis, 11 patients with concurrent malignancies, and 11 patients with other malignancies after their LCH diagnosis. Solid tumors comprised 74% of the malignancies, lymphomas comprised 17% of the cases, and hematologic malignancies comprised 9% of the cases. Seventy-one percent of the patients were smokers.[39] These results are in contrast to an earlier study that was based on a literature review and institutional surveys that reported a higher incidence of lymphomas concurrent with the LCH diagnosis.[40]
The association between LCH and malignancy occurs more frequently than would be expected by chance, based on questionnaires sent to investigators in the Histiocyte Society and a literature review. In one publication, LCH-malignancy cases were collected between 1991 and 2015. A total of 285 LCH-malignancies were seen in 270 patients. In 154 adults with LCH, solid tumors were reported in 61 patients (39.6%), lymphomas in 56 patients (36.4%), and leukemias and myeloproliferative disorders in 37 patients (24.0%). Thyroid malignancy was also seen with some frequency. In adults, LCH and malignancy occurred concurrently in 69 patients (44.8%).[41]
A review of Surveillance, Epidemiology, and End Results (SEER) Program data for subsequent malignancies in 456 adults with LCH found 16 cases.[42] There were two cases of non-Hodgkin lymphoma, two cases of myelodysplastic neoplasms, three cases of breast cancer, three cases of lung cancer, and one case each of colorectal cancer, thyroid cancer, vulvar cancer, meningioma, and adenocarcinoma, not otherwise specified.
A study of 156 adults with LCH reported on the relationship of LCH with the BRAF V600E variant and secondary primary malignancies.[43] Patients with LCH and the variant had a 17.3% incidence of second primary malignancies, compared with 4.1% for patients without the variant. The standardized incidence ratio (SIR) was 5.72 for second malignancies in patients with LCH, compared with 1.7 in age-matched adults. Unlike children with LCH, there was no correlation with the extent of the disease or progression-free survival in adults with BRAF V600E variants.
Diagnostic Evaluation
Positron emission tomography (PET) scans are the most sensitive modality for finding affected sites and are done to diagnose people with LCH.[1,44] MRI of the brain is indicated for patients with pituitary-associated symptoms and those with evidence of neurodegeneration. Spine MRIs are indicated for people with vertebral pain or lower motor neuropathy.
Treatment of Adult LCH
Treatment options for adult LCH
The lack of clinical trials limits the ability to make evidence-based recommendations for adult patients with LCH.
Many investigators have previously recommended treatment according to the guidelines for childhood LCH. It is unclear, however, whether adult LCH responds as well as the childhood form of the disease. In addition, the drugs used in the treatment of children are not as well tolerated when used in adults. Excessive neurological toxicity from vinblastine, for example, prompted closure of the LCH-A1 trial. BRAF and MEK inhibitors are increasingly used as initial treatment for many adults.[1] For more information, see the Targeted therapies for the treatment of single-system and multisystem disease section.
An international expert consensus panel has proposed a treatment algorithm for adult patients and is summarized below.[1]
- Bone-only LCH: Treatment includes curettage, bisphosphonates, oral methotrexate, or hydroxyurea. Radiation therapy may be used for patients with progressive disease and fewer than three lesions. Patients with more than three lesions may receive chemotherapy (cladribine, cytarabine, or others) or MAPK inhibitors.
- Skin-only LCH: Treatment includes hydroxyurea, oral methotrexate, thalidomide or lenalidomide, or topical therapy.
- Single-system pulmonary LCH: Treatment is smoking cessation. Patients with progressive disease receive chemotherapy with similar drugs used for bone LCH.
- Multisystem LCH: Treatment includes chemotherapy with similar drugs used for bone LCH.
- Critical organ involvement (bone marrow, spleen, liver, brain): Patients with LCH and BRAF V600E variants receive a BRAF inhibitor. For patients without BRAF V600E variants, a MEK inhibitor may be used if tests are positive for MAPK variants. If no variants are found, patients receive chemotherapy based on the regimen used for bone LCH.
Treatment of pulmonary LCH
It is difficult to judge the effectiveness of various treatments for pulmonary LCH because patients can recover spontaneously or have stable disease without treatment.
Treatment options for adult patients with pulmonary LCH include the following:
- Smoking cessation. Smoking cessation is mandatory because of the apparent causal effect of smoking in pulmonary LCH.[45] Most adult patients with LCH have gradual disease progression with continued smoking. The disease may regress or progress with the cessation of smoking.[46] A study of 27 patients with pulmonary LCH observed that 52% of patients improved after a mean follow-up of 14 months. Most patients improved with smoking cessation, and some patients improved with steroid treatment. Four patients (15%) had stable disease at a mean follow-up of 26 months, and nine patients (33%) demonstrated disease progression during the mean follow-up of 22 months.[30][Level of evidence C3]
- Steroid therapy. It is not known whether steroid therapy is efficacious in the treatment of adult pulmonary LCH because reported case series did not control for smoking cessation.[45]
- Chemotherapy. Some patients have been reported to respond to treatment with cladribine or cytarabine.[45,47]; [15][Level of evidence C2]
- Lung transplant. Lung transplant may be necessary for adults with extensive pulmonary destruction from LCH.[22] One multicenter study reported a survival rate of 54% at 10 years posttransplant, with 20% of patients having recurrent LCH that did not impact survival. Longer follow-up of these patients is needed.[22] Another study confirmed a survival rate of approximately 50% at 10 years and improved hemodynamic changes associated with pulmonary arterial hypertension therapies, without oxygen worsening or pulmonary edema.[48]
The best strategy for follow-up of pulmonary LCH includes physical examination, chest radiographs, lung function tests, and high-resolution CT scans.[49]
Treatment of bone LCH
Treatment options for adult patients with bone LCH include the following:
- Curettage followed by observation, with or without intralesional corticosteroids. As in children, adults with single-bone lesions should undergo curettage of the lesion followed by observation, with or without intralesional corticosteroids.[50] Extensive or radical surgery leading to loss of function and disfigurement is contraindicated at any site, including the teeth or jaw bones.
- Systemic chemotherapy. Systemic chemotherapy causes bone lesions to regress. A variety of chemotherapy regimens, including cytarabine and cladribine, have been used in the treatment of a relatively limited number of patients.[51,52] For more information, see the Chemotherapy and radiation therapy for the treatment of other single-system disease and multisystem disease section.
-
Low-dose radiation therapy. For patients who do not respond to chemotherapy, low-dose radiation therapy may be indicated and should be attempted before any radical surgery. Radiation therapy is also indicated for impending neurological deficits from vertebral body lesions or visual problems from orbital lesions. Two series and a study have reported the following:
- A German cooperative radiation therapy group reported on a series of 98 adult patients with LCH. Most of the patients (60 of 98) had only bone lesions and 24 had multisystem disease including bone, who were treated with radiation therapy.[53][Level of evidence C3] Of 89 evaluable patients, 77% achieved a complete remission, 9% developed an infield recurrence, and 15.7% (14 of 89) experienced a progression outside the radiation field(s).
- A retrospective analysis of 80 patients treated with radiation therapy alone reported a complete remission rate of 77% and a partial remission rate of 12.5%. The long-term control rate was 80% in adults. No adverse late effects were reported.[54][Level of evidence C3]
- A single-institution study included 39 patients with LCH (age range, 1.5–67 years; 24 patients aged >18 years) who received radiation therapy to 46 lesions. The study reported no local recurrences in the 31 bony sites. In comparison, the 3-year freedom from local failure rate was 63% in the 15 nonbone lesions (95% confidence interval, 32%–83%; P = .0008).[55]
- Bisphosphonate therapy. Case reports and case series have described the successful use of bisphosphonates, both intravenous pamidronate and oral zoledronate, in controlling severe bone pain in patients with multiple osteolytic LCH bone lesions.[56,57,58] A multi-institutional review of bisphosphonate therapy in children and adults with LCH found that most adult patients were given oral zoledronic acid, and most pediatric patients were given pamidronate.[59][Level of evidence C3] Because of the increased toxicity of chemotherapy in adults, bisphosphonate therapy could be used before chemotherapy in multifocal bone disease. Response of other organs, such as skin and soft tissue, to bisphosphonate therapy has been reported.[60]
- Anti-inflammatory agents with trofosfamide. Another approach using anti-inflammatory agents (pioglitazone and rofecoxib) coupled with trofosfamide in a specific timed sequence was successful in two patients who had disease resistant to standard chemotherapy treatment.[61][Level of evidence C3]
Treatment of single-system skin disease
Treatment options for adult patients with single-system skin disease include the following:
- Surgical excision. Localized lesions are rarely treated by surgical excision. Mutilating surgery, including hemivulvectomy, should be avoided unless the disease is refractory to all available therapy.
-
Topical therapy. Topical therapies are described in greater detail in the childhood isolated skin involvement section and include the following:
- Topical or intralesional corticosteroid.
- Topical tacrolimus.[62][Level of evidence C2]
- Topical imiquimod.[63,64][Level of evidence C3]
- Psoralen and long-wave ultraviolet A radiation (PUVA) and UVB. Therapies such as PUVA/UVB may be more useful in adults because long-term toxicity may be reduced.[62,65][Level of evidence C3]
-
Systemic therapy. Systemic therapy for severe skin LCH includes oral methotrexate, hydroxyurea, oral thalidomide, oral interferon-alpha, or combinations of interferon and thalidomide.[66,67,68][Level of evidence C3] Interferon and thalidomide are also used to treat chronic skin LCH in adults.[69][Level of evidence C3] Recurrences are possible after treatment is stopped but lesions usually respond to re-treatment.
Oral isotretinoin has induced remissions in some adult patients with refractory skin LCH.[70][Level of evidence C3]
Chemotherapy and radiation therapy for the treatment of other single-system disease and multisystem disease
Evidence (chemotherapy for the treatment of other single-system disease [not mentioned above] and multisystem disease):
- A single-center, retrospective review of 58 adult patients with LCH reported on the efficacy and toxicities of treatment with vinblastine/prednisone, cladribine, and cytarabine.[51][Level of evidence C3]
- Patients treated with vinblastine/prednisone had the worst outcome, with 84% of patients not responding within 6 weeks or relapsing within a year.
- The no-response/relapse rate was 59% for patients treated with cladribine and 21% for patients treated with cytarabine.
- Grade 3 or 4 neurological toxic effects occurred in 75% of patients treated with vinblastine.
- Grade 3 or 4 neutropenia occurred in 37% of patients treated with cladribine and in 20% of patients who received cytarabine.
- One report evaluated adult patients who were treated with either vindesine and prednisone or cyclophosphamide, etoposide, vindesine, and prednisone.[71][Level of evidence C2]
- More than 70% of patients relapsed with either regimen.
- Etoposide has been used with some success in adult patients with single-system and multisystem LCH.
- Minimal toxicity was reported with the use of prolonged oral etoposide in adults with skin LCH, while 3-day courses of intravenous etoposide (100 mg/m2 /day) induced complete remission in a small number of patients with resistant single-system and multisystem disease.[72][Level of evidence C3]
- Another study at the same center found that azathioprine was the most successful drug for localized disease in adults, with the addition of etoposide for refractory and multisystem disease.[73][Level of evidence C3]
- Cladribine is effective for adults with skin, bone, lymph node, and probably pulmonary and CNS disease.[74,75][Level of evidence C3]; [52]
- In a retrospective multicenter study, 23 patients who had at least one previous therapy were treated with cladribine, using various dosing and treatment schedules.[76][Level of evidence C2] The overall response rate was 91%, and the complete response rate was 50%. A literature review identified an additional 48 patients who were treated with cladribine. The pooled analysis confirmed these results.
- A study reported on 38 patients with newly diagnosed or relapsed/refractory LCH treated with one to nine cycles of cladribine.[1][Level of evidence C2] The overall response rate was 79%. The complete response rate was 26%, and the partial response rate was 53%.[52]
- An adult lymphoma treatment regimen of methotrexate, doxorubicin, cyclophosphamide, vincristine, prednisone, and bleomycin (MACOP-B) was used in 11 patients.[77]
- The overall response rate was 100%, and the progression-free survival rate was 64%.
- Methotrexate and cytarabine were given to 83 patients with newly diagnosed lung (68%), liver (28%), spleen (13%), or nonpituitary (4%) LCH.[78]
- The objective response rate was 88%, and one-third of the patients progressed after 3 years.
- There was a high rate of grades 3 to 4 neutropenia, with nearly one-half of patients developing fevers.
- One-third of patients had grades 3 to 4 thrombocytopenia.
- In a study of 61 adult patients with LCH who were treated with subcutaneous cytarabine, the following results were reported:[79]
- The estimated 3-year event-free survival (EFS) rate was 58.5%.
- Poor prognostic factors for EFS included three or more involved organs and baseline lung involvement.
- Grades 3 to 4 neutropenia occurred in 27.9% of patients.
- Of the 61 patients, 47 completed 12 months of treatment, and 14 left the study (6 had a poor response to therapy and 8 decided to withdraw).
Radiation therapy. A report of stereotactic radiosurgery for the treatment of adult patients with pituitary LCH showed efficacy in reducing the masses.[80]
Targeted therapies for the treatment of single-system and multisystem disease
Early reports on the use of targeted therapies for adult patients with low-risk or high-risk LCH sites include the following:
-
MAP2K/ERK pathway inhibitors. The finding that most patients with LCH have BRAF or other RAS pathway variants led to several reports of good responses to vemurafenib, a BRAF V600E inhibitor, in adult patients with LCH and severe cutaneous LCH.[81,82,83,84,85][Level of evidence C3]
Of four patients with LCH who were treated with vemurafenib on the VE-BASKET (NCT01524978) trial, one patient had a complete response and three patients had partial responses.[84][Level of evidence C3] One patient with LCH who was treated with vemurafenib had improvement in ataxia.[84][Level of evidence C3]
One series reported on six patients who were treated with BRAF inhibitors as initial therapy.[86] Five patients had multisystem disease, and one patient had bone-only LCH. There were two complete responses, three partial responses, and one stable disease after 4 to 27 months of treatment.
A proof-of-concept clinical trial of cobimetinib, an oral inhibitor of MEK1 and MEK2, was carried out in 18 adult patients with various histiocytoses, including histiocytic sarcomas. Patients were treated regardless of genomic findings. Responses were seen in patients with ARAF, BRAF, NRAS, KRAS, MAP2K1, and MAP2K2 variants. The overall response rate was 89%, with responses being durable. At 1 year, 94% of patients remained progression free.[83][Level of evidence C2]
Early results of targeted inhibitor therapy are encouraging, but many questions remain, particularly the optimal duration of therapy and the reactivation rate after therapy is discontinued. A BRAF inhibitor in combination with a MEK inhibitor have been shown to be effective in patients with melanoma who have BRAF variants (with reduced toxicity). This combination may also be effective in patients with LCH, but it is generally not used for patients with histiocytic diseases.[81][Level of evidence C3] A number of clinical trials of BRAF and other RAS pathway inhibitors in adults and children with LCH are ongoing.
- Other targeted therapies. A case report suggests some benefit to treating neurodegenerative CNS LCH disease with infliximab, a tumor necrosis factor (TNF)-alpha inhibitor.[87][Level of evidence C3] However, the TNF inhibitors infliximab and etanercept have limited ability to cross the blood-brain barrier. Thalidomide, which also has anti-TNF activity, has been effective in adults with skin and bone LCH.[66,88][Level of evidence C3]; [88,89]
Treatment options under clinical evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- NCT04079179 (Cobimetinib for the Treatment of Refractory LCH): This study is open to children or adults with relapsed or refractory LCH or other newly diagnosed, relapsed, or refractory histiocytic disorders.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
- Goyal G, Tazi A, Go RS, et al.: International expert consensus recommendations for the diagnosis and treatment of Langerhans cell histiocytosis in adults. Blood 139 (17): 2601-2621, 2022.
- Goyal G, Acosta-Medina AA, Abeykoon JP, et al.: Long-term outcomes among adults with Langerhans cell histiocytosis. Blood Adv 7 (21): 6568-6578, 2023.
- Liu H, Stiller CA, Crooks CJ, et al.: Incidence, prevalence and survival in patients with Langerhans cell histiocytosis: A national registry study from England, 2013-2019. Br J Haematol 199 (5): 728-738, 2022.
- Tazi A, Soler P, Hance AJ: Adult pulmonary Langerhans' cell histiocytosis. Thorax 55 (5): 405-16, 2000.
- Vassallo R, Ryu JH, Colby TV, et al.: Pulmonary Langerhans'-cell histiocytosis. N Engl J Med 342 (26): 1969-78, 2000.
- Yousem SA, Colby TV, Chen YY, et al.: Pulmonary Langerhans' cell histiocytosis: molecular analysis of clonality. Am J Surg Pathol 25 (5): 630-6, 2001.
- Roden AC, Hu X, Kip S, et al.: BRAF V600E expression in Langerhans cell histiocytosis: clinical and immunohistochemical study on 25 pulmonary and 54 extrapulmonary cases. Am J Surg Pathol 38 (4): 548-51, 2014.
- Baumgartner I, von Hochstetter A, Baumert B, et al.: Langerhans'-cell histiocytosis in adults. Med Pediatr Oncol 28 (1): 9-14, 1997.
- Kaltsas GA, Powles TB, Evanson J, et al.: Hypothalamo-pituitary abnormalities in adult patients with langerhans cell histiocytosis: clinical, endocrinological, and radiological features and response to treatment. J Clin Endocrinol Metab 85 (4): 1370-6, 2000.
- Aricò M, Girschikofsky M, Généreau T, et al.: Langerhans cell histiocytosis in adults. Report from the International Registry of the Histiocyte Society. Eur J Cancer 39 (16): 2341-8, 2003.
- Götz G, Fichter J: Langerhans'-cell histiocytosis in 58 adults. Eur J Med Res 9 (11): 510-4, 2004.
- Slater JM, Swarm OJ: Eosinophilic granuloma of bone. Med Pediatr Oncol 8 (2): 151-64, 1980.
- Vassallo R, Ryu JH, Schroeder DR, et al.: Clinical outcomes of pulmonary Langerhans'-cell histiocytosis in adults. N Engl J Med 346 (7): 484-90, 2002.
- Schönfeld N, Frank W, Wenig S, et al.: Clinical and radiologic features, lung function and therapeutic results in pulmonary histiocytosis X. Respiration 60 (1): 38-44, 1993.
- Miao HL, Zhao AL, Duan MH, et al.: Clinical presentation and prognostic analysis of adult patients with Langerhans cell histiocytosis with pulmonary involvement. BMC Cancer 20 (1): 911, 2020.
- Travis WD, Borok Z, Roum JH, et al.: Pulmonary Langerhans cell granulomatosis (histiocytosis X). A clinicopathologic study of 48 cases. Am J Surg Pathol 17 (10): 971-86, 1993.
- Tazi A, Moreau J, Bergeron A, et al.: Evidence that Langerhans cells in adult pulmonary Langerhans cell histiocytosis are mature dendritic cells: importance of the cytokine microenvironment. J Immunol 163 (6): 3511-5, 1999.
- Baqir M, Vassallo R, Maldonado F, et al.: Utility of bronchoscopy in pulmonary Langerhans cell histiocytosis. J Bronchology Interv Pulmonol 20 (4): 309-12, 2013.
- Kamionek M, Ahmadi Moghaddam P, Sakhdari A, et al.: Mutually exclusive extracellular signal-regulated kinase pathway mutations are present in different stages of multi-focal pulmonary Langerhans cell histiocytosis supporting clonal nature of the disease. Histopathology 69 (3): 499-509, 2016.
- Benattia A, Bugnet E, Walter-Petrich A, et al.: Long-term outcomes of adult pulmonary Langerhans cell histiocytosis: a prospective cohort. Eur Respir J 59 (5): , 2022.
- Delobbe A, Durieu J, Duhamel A, et al.: Determinants of survival in pulmonary Langerhans' cell granulomatosis (histiocytosis X). Groupe d'Etude en Pathologie Interstitielle de la Société de Pathologie Thoracique du Nord. Eur Respir J 9 (10): 2002-6, 1996.
- Dauriat G, Mal H, Thabut G, et al.: Lung transplantation for pulmonary langerhans' cell histiocytosis: a multicenter analysis. Transplantation 81 (5): 746-50, 2006.
- Chaulagain CP: Pulmonary langerhans' cell histiocytosis. Am J Med 122 (11): e5-6, 2009.
- Lin MW, Chang YL, Lee YC, et al.: Pulmonary Langerhans cell histiocytosis. Lung 187 (4): 261-2, 2009.
- Tazi A, Hiltermann J, Vassallo R: Adult lung histiocytosis. In: Weitzman S, Egeler R M, eds.: Histiocytic Disorders of Children and Adults. Cambridge, United Kingdom: Cambridge University Press, 2005, pp 187-207.
- Tazi A, de Margerie C, Naccache JM, et al.: The natural history of adult pulmonary Langerhans cell histiocytosis: a prospective multicentre study. Orphanet J Rare Dis 10: 30, 2015.
- Crausman RS, Jennings CA, Tuder RM, et al.: Pulmonary histiocytosis X: pulmonary function and exercise pathophysiology. Am J Respir Crit Care Med 153 (1): 426-35, 1996.
- Diette GB, Scatarige JC, Haponik EF, et al.: Do high-resolution CT findings of usual interstitial pneumonitis obviate lung biopsy? Views of pulmonologists. Respiration 72 (2): 134-41, 2005 Mar-Apr.
- Soler P, Bergeron A, Kambouchner M, et al.: Is high-resolution computed tomography a reliable tool to predict the histopathological activity of pulmonary Langerhans cell histiocytosis? Am J Respir Crit Care Med 162 (1): 264-70, 2000.
- Kim HJ, Lee KS, Johkoh T, et al.: Pulmonary Langerhans cell histiocytosis in adults: high-resolution CT-pathology comparisons and evolutional changes at CT. Eur Radiol 21 (7): 1406-15, 2011.
- Abdallah M, Généreau T, Donadieu J, et al.: Langerhans' cell histiocytosis of the liver in adults. Clin Res Hepatol Gastroenterol 35 (6-7): 475-81, 2011.
- Makras P, Alexandraki KI, Chrousos GP, et al.: Endocrine manifestations in Langerhans cell histiocytosis. Trends Endocrinol Metab 18 (6): 252-7, 2007.
- Sagna Y, Courtillot C, Drabo JY, et al.: Endocrine manifestations in a cohort of 63 adulthood and childhood onset patients with Langerhans cell histiocytosis. Eur J Endocrinol 181 (3): 275-285, 2019.
- Goyal G, Hu M, Young JR, et al.: Adult Langerhans cell histiocytosis: a contemporary single-institution series of 186 patients. [Abstract] J Clin Oncol 37 (Suppl 15): A-7018, 2019. Also available online. Last accessed February 27, 2024.
- Kim HK, Park CJ, Jang S, et al.: Bone marrow involvement of Langerhans cell histiocytosis: immunohistochemical evaluation of bone marrow for CD1a, Langerin, and S100 expression. Histopathology 65 (6): 742-8, 2014.
- Singhi AD, Montgomery EA: Gastrointestinal tract langerhans cell histiocytosis: A clinicopathologic study of 12 patients. Am J Surg Pathol 35 (2): 305-10, 2011.
- Chen CY, Wu MH, Huang SF, et al.: Langerhans' cell histiocytosis presenting with a para-aortic lesion and heart failure. J Formos Med Assoc 100 (2): 127-30, 2001.
- Howarth DM, Gilchrist GS, Mullan BP, et al.: Langerhans cell histiocytosis: diagnosis, natural history, management, and outcome. Cancer 85 (10): 2278-90, 1999.
- Ma J, Laird JH, Chau KW, et al.: Langerhans cell histiocytosis in adults is associated with a high prevalence of hematologic and solid malignancies. Cancer Med 8 (1): 58-66, 2019.
- Egeler RM, Neglia JP, Puccetti DM, et al.: Association of Langerhans cell histiocytosis with malignant neoplasms. Cancer 71 (3): 865-73, 1993.
- Bagnasco F, Zimmermann SY, Egeler RM, et al.: Langerhans cell histiocytosis and associated malignancies: A retrospective analysis of 270 patients. Eur J Cancer 172: 138-145, 2022.
- Goyal G, Parikh R, Richman J, et al.: Spectrum of second primary malignancies and cause-specific mortality in pediatric and adult langerhans cell histiocytosis. Leuk Res 126: 107032, 2023.
- Acosta-Medina AA, Kemps PG, Zondag TCE, et al.: BRAF V600E is associated with higher incidence of second cancers in adults with Langerhans cell histiocytosis. Blood 142 (18): 1570-1575, 2023.
- An R, Ma X, Wang Y: The value of 18F-FDG PET/CT in Langerhans cell histiocytosis. Ann Nucl Med 38 (3): 238-245, 2024.
- Tazi A: Adult pulmonary Langerhans' cell histiocytosis. Eur Respir J 27 (6): 1272-85, 2006.
- Mogulkoc N, Veral A, Bishop PW, et al.: Pulmonary Langerhans' cell histiocytosis: radiologic resolution following smoking cessation. Chest 115 (5): 1452-5, 1999.
- Lorillon G, Tazi A: How I manage pulmonary Langerhans cell histiocytosis. Eur Respir Rev 26 (145): , 2017.
- Le Pavec J, Lorillon G, Jaïs X, et al.: Pulmonary Langerhans cell histiocytosis-associated pulmonary hypertension: clinical characteristics and impact of pulmonary arterial hypertension therapies. Chest 142 (5): 1150-1157, 2012.
- Abbritti M, Mazzei MA, Bargagli E, et al.: Utility of spiral CAT scan in the follow-up of patients with pulmonary Langerhans cell histiocytosis. Eur J Radiol 81 (8): 1907-12, 2012.
- Christopher Z, Binitie O, Henderson-Jackson E, et al.: Langerhans cell histiocytosis of bone in an adult: A case report. Radiol Case Rep 13 (2): 310-314, 2018.
- Cantu MA, Lupo PJ, Bilgi M, et al.: Optimal therapy for adults with Langerhans cell histiocytosis bone lesions. PLoS One 7 (8): e43257, 2012.
- Goyal G, Abeykoon JP, Hu M, et al.: Single-agent cladribine as an effective front-line therapy for adults with Langerhans cell histiocytosis. Am J Hematol 96 (5): E146-E150, 2021.
- Olschewski T, Seegenschmiedt MH: Radiotherapy of Langerhans' Cell Histiocytosis : Results and Implications of a National Patterns-of-Care Study. Strahlenther Onkol 182 (11): 629-34, 2006.
- Kriz J, Eich HT, Bruns F, et al.: Radiotherapy in langerhans cell histiocytosis - a rare indication in a rare disease. Radiat Oncol 8: 233, 2013.
- Laird J, Ma J, Chau K, et al.: Outcome After Radiation Therapy for Langerhans Cell Histiocytosis Is Dependent on Site of Involvement. Int J Radiat Oncol Biol Phys 100 (3): 670-678, 2018.
- Arzoo K, Sadeghi S, Pullarkat V: Pamidronate for bone pain from osteolytic lesions in Langerhans'-cell histiocytosis. N Engl J Med 345 (3): 225, 2001.
- Farran RP, Zaretski E, Egeler RM: Treatment of Langerhans cell histiocytosis with pamidronate. J Pediatr Hematol Oncol 23 (1): 54-6, 2001.
- Brown RE: Bisphosphonates as antialveolar macrophage therapy in pulmonary langerhans cell histiocytosis? Med Pediatr Oncol 36 (6): 641-3, 2001.
- Chellapandian D, Makras P, Kaltsas G, et al.: Bisphosphonates in Langerhans Cell Histiocytosis: An International Retrospective Case Series. Mediterr J Hematol Infect Dis 8 (1): e2016033, 2016.
- Morimoto A, Shioda Y, Imamura T, et al.: Nationwide survey of bisphosphonate therapy for children with reactivated Langerhans cell histiocytosis in Japan. Pediatr Blood Cancer 56 (1): 110-5, 2011.
- Reichle A, Vogt T, Kunz-Schughart L, et al.: Anti-inflammatory and angiostatic therapy in chemorefractory multisystem Langerhans' cell histiocytosis of adults. Br J Haematol 128 (5): 730-2, 2005.
- Rieker J, Hengge U, Ruzicka T, et al.: [Multifocal facial eosinophilic granuloma: successful treatment with topical tacrolimus]. Hautarzt 57 (4): 324-6, 2006.
- O'Kane D, Jenkinson H, Carson J: Langerhans cell histiocytosis associated with breast carcinoma successfully treated with topical imiquimod. Clin Exp Dermatol 34 (8): e829-32, 2009.
- Taverna JA, Stefanato CM, Wax FD, et al.: Adult cutaneous Langerhans cell histiocytosis responsive to topical imiquimod. J Am Acad Dermatol 54 (5): 911-3, 2006.
- Vogel CA, Aughenbaugh W, Sharata H: Excimer laser as adjuvant therapy for adult cutaneous Langerhans cell histiocytosis. Arch Dermatol 144 (10): 1287-90, 2008.
- McClain KL, Kozinetz CA: A phase II trial using thalidomide for Langerhans cell histiocytosis. Pediatr Blood Cancer 48 (1): 44-9, 2007.
- Steen AE, Steen KH, Bauer R, et al.: Successful treatment of cutaneous Langerhans cell histiocytosis with low-dose methotrexate. Br J Dermatol 145 (1): 137-40, 2001.
- Zinn DJ, Grimes AB, Lin H, et al.: Hydroxyurea: a new old therapy for Langerhans cell histiocytosis. Blood 128 (20): 2462-2465, 2016.
- Chang SE, Koh GJ, Choi JH, et al.: Widespread skin-limited adult Langerhans cell histiocytosis: long-term follow-up with good response to interferon alpha. Clin Exp Dermatol 27 (2): 135-7, 2002.
- Tsambaos D, Georgiou S, Kapranos N, et al.: Langerhans' cell histiocytosis: complete remission after oral isotretinoin therapy. Acta Derm Venereol 75 (1): 62-4, 1995.
- Duan MH, Han X, Li J, et al.: Comparison of vindesine and prednisone and cyclophosphamide, etoposide, vindesine, and prednisone as first-line treatment for adult Langerhans cell histiocytosis: A single-center retrospective study. Leuk Res 42: 43-6, 2016.
- Tsele E, Thomas DM, Chu AC: Treatment of adult Langerhans cell histiocytosis with etoposide. J Am Acad Dermatol 27 (1): 61-4, 1992.
- Chu T: Langerhans cell histiocytosis. Australas J Dermatol 42 (4): 237-42, 2001.
- Saven A, Foon KA, Piro LD: 2-Chlorodeoxyadenosine-induced complete remissions in Langerhans-cell histiocytosis. Ann Intern Med 121 (6): 430-2, 1994.
- Pardanani A, Phyliky RL, Li CY, et al.: 2-Chlorodeoxyadenosine therapy for disseminated Langerhans cell histiocytosis. Mayo Clin Proc 78 (3): 301-6, 2003.
- Néel A, Artifoni M, Fontenoy AM, et al.: Long-term efficacy and safety of 2CdA (cladribine) in extra-pulmonary adult-onset Langerhans cell histiocytosis: analysis of 23 cases from the French Histiocytosis Group and systematic literature review. Br J Haematol 189 (5): 869-878, 2020.
- Derenzini E, Stefoni V, Pellegrini C, et al.: High efficacy of the MACOP-B regimen in the treatment of adult Langerhans cell histiocytosis, a 20 year experience. BMC Cancer 15: 879, 2015.
- Cao XX, Li J, Zhao AL, et al.: Methotrexate and cytarabine for adult patients with newly diagnosed Langerhans cell histiocytosis: A single arm, single center, prospective phase 2 study. Am J Hematol 95 (9): E235-E238, 2020.
- Chang L, Lang M, Lin H, et al.: Phase 2 study using low dose cytarabine for adult patients with newly diagnosed Langerhans cell histiocytosis. Leukemia 38 (4): 803-809, 2024.
- Hong WC, Murovic JA, Gibbs I, et al.: Pituitary stalk Langerhans cell histiocytosis treated with CyberKnife radiosurgery. Clin Neurol Neurosurg 115 (5): 573-7, 2013.
- Haroche J, Cohen-Aubart F, Emile JF, et al.: Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood 121 (9): 1495-500, 2013.
- Charles J, Beani JC, Fiandrino G, et al.: Major response to vemurafenib in patient with severe cutaneous Langerhans cell histiocytosis harboring BRAF V600E mutation. J Am Acad Dermatol 71 (3): e97-9, 2014.
- Diamond EL, Durham BH, Ulaner GA, et al.: Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature 567 (7749): 521-524, 2019.
- Diamond EL, Subbiah V, Lockhart AC, et al.: Vemurafenib for BRAF V600-Mutant Erdheim-Chester Disease and Langerhans Cell Histiocytosis: Analysis of Data From the Histology-Independent, Phase 2, Open-label VE-BASKET Study. JAMA Oncol 4 (3): 384-388, 2018.
- Hyman DM, Puzanov I, Subbiah V, et al.: Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N Engl J Med 373 (8): 726-36, 2015.
- Hazim AZ, Ruan GJ, Ravindran A, et al.: Efficacy of BRAF-Inhibitor Therapy in BRAFV600E -Mutated Adult Langerhans Cell Histiocytosis. Oncologist 25 (12): 1001-1004, 2020.
- Chohan G, Barnett Y, Gibson J, et al.: Langerhans cell histiocytosis with refractory central nervous system involvement responsive to infliximab. J Neurol Neurosurg Psychiatry 83 (5): 573-5, 2012.
- Sander CS, Kaatz M, Elsner P: Successful treatment of cutaneous langerhans cell histiocytosis with thalidomide. Dermatology 208 (2): 149-52, 2004.
- Crickx E, Bouaziz JD, Lorillon G, et al.: Clinical Spectrum, Quality of Life, BRAF Mutation Status and Treatment of Skin Involvement in Adult Langerhans Cell Histiocytosis. Acta Derm Venereol 97 (7): 838-842, 2017.
Latest Updates to This Summary (01 / 06 / 2025)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Editorial changes were made to this summary.
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of childhood and adult Langerhans cell histiocytosis. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Langerhans Cell Histiocytosis Treatment are:
- Louis S. Constine, MD (James P. Wilmot Cancer Center at University of Rochester Medical Center)
- Thomas G. Gross, MD, PhD (National Cancer Institute)
- Michael Jeng, MD (Stanford Medicine Children's Health)
- Kenneth L. McClain, MD, PhD (Texas Children's Cancer Center and Hematology Service at Texas Children's Hospital)
- Carlos Rodriguez-Galindo, MD (St. Jude Children's Research Hospital)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as "NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary]."
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Langerhans Cell Histiocytosis Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/langerhans/hp/langerhans-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389240]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either "standard" or "under clinical evaluation." These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website's Email Us.
Last Revised: 2025-01-06

